Junge Patientin mit AV-Ueberleitungsstörungen und polymorphen VES - Rolf Handschin, Christian Sticherling, Annina Vischer Cardiolunch 29.10.2020
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

Junge Patientin mit AV-Ueberleitungsstörungen
und polymorphen VES
Rolf Handschin, Christian Sticherling, Annina Vischer
Cardiolunch 29.10.2020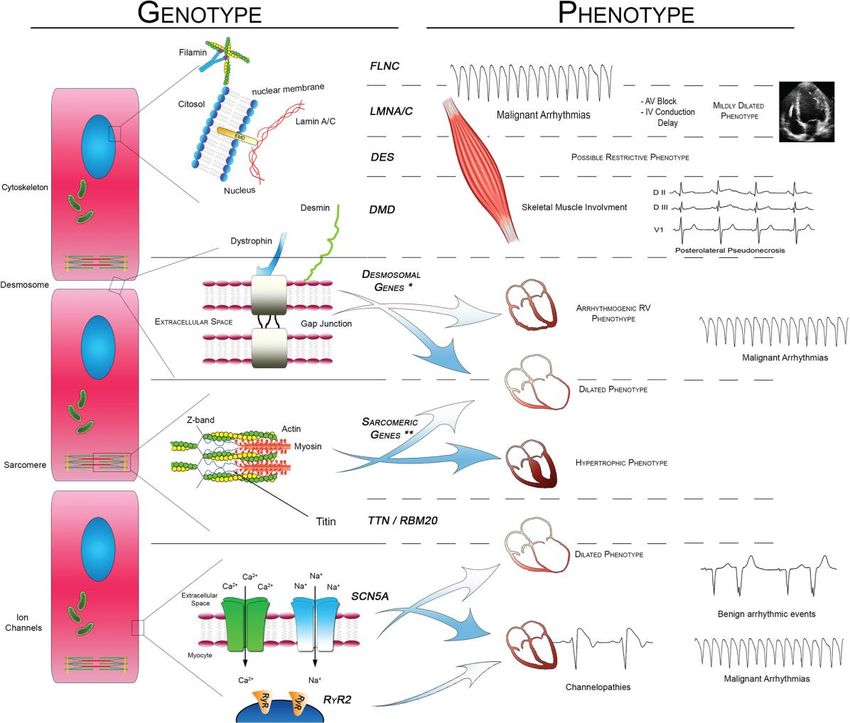
Initiale Präsentation KSBL - 1 41-Jährige Patientin stellt sich notfallmässig im KSBL-Bruderholz 12/2019 vor Anamnese: seit 10 Jahren bekannte Palpitationen, jetzt verstärkt („Herzstolpern“). Assoziiert mit Schwindel und thorakalem Druckgefühl. Keine Synkopen. PA: Kardiologische Abklärungen 2007 (USB) und später Zürich: VES FA: Mutter 48-jährig mit Herzschrittmacher und dilatativer Kardiomyopathie verstorben (ZH, Akten nicht mehr vorhanden). Ein Halbbruder (gemeinsame Mutter), zwei Kinder im Primarschulalter, bisher alle herzgesund. Klinik: Normotensiv, normokard, afebril und normoxäm. Kardialer Status bis auf ES unauffällig, internistisch sonst unauffällig.
Initiales EKG KSBL
Initiale Präsentation KSBL - 2 Während ICU Überwachung häufig AV Block Grad II Typ 1 (Wenckebach) Labor: hs-Troponin T stets leicht erhöht (15-16 ng/l), keine Entzündungszeichen, Borrelienserologie unauffällig Transthorakale Echokardiographie: keine pathologische Befunde cMRI: leicht dilatierter linker Ventrikel mit leicht eingeschränkter systolischer Funktion, midwall streak bei late gadolinium Sequenzen
Kardiale Magnetresonanztomographie KSBL

Initiale Präsentation KSBL - 3 Fahrradergometrie: nur noch AV Block Grad I, Abnahme VES unter Last 24-Stunden EKG ambulant: Polymorphe VES, VES Last 6.8%. Keine Kammertachykardien. AV Block Grad I bis II Typ 1

Beurteilung KSBL
Diagnosen:
AV-Block 2. Grades Typ 1 (Wenckebach), ED 12/2019
symptomatisch mit Herzstolpern/Schwindel
Borrelien-Serologie unauffällig
Ventrikuläre Extrasystolie, ED 01/2007
symptomatisch mit Herzstolpern
polymorph, häufigste Morphologie: inferiore Achse und RSB-Bild
VES-Last 6.75 %
Möglicherweise familiäre dilatative Kardiomyopathie, ED 12/2019
leicht dilatierter linker Ventrikel mit leicht eingeschräkter Auswurffraktion
keine Hinweise für eine infiltrative/entzündliche Kardiomyopathie -
speziell Sarkoidose (CMR)
midwall streak im late Gadolinium enhancement (DCM-Kriterium)
stabil leicht erhöhtes hs Troponin T (15-16ng/l)
Mutter 48-jährig mit Herzschrittmacher und dilatativer Kardiomyopathie
verstorben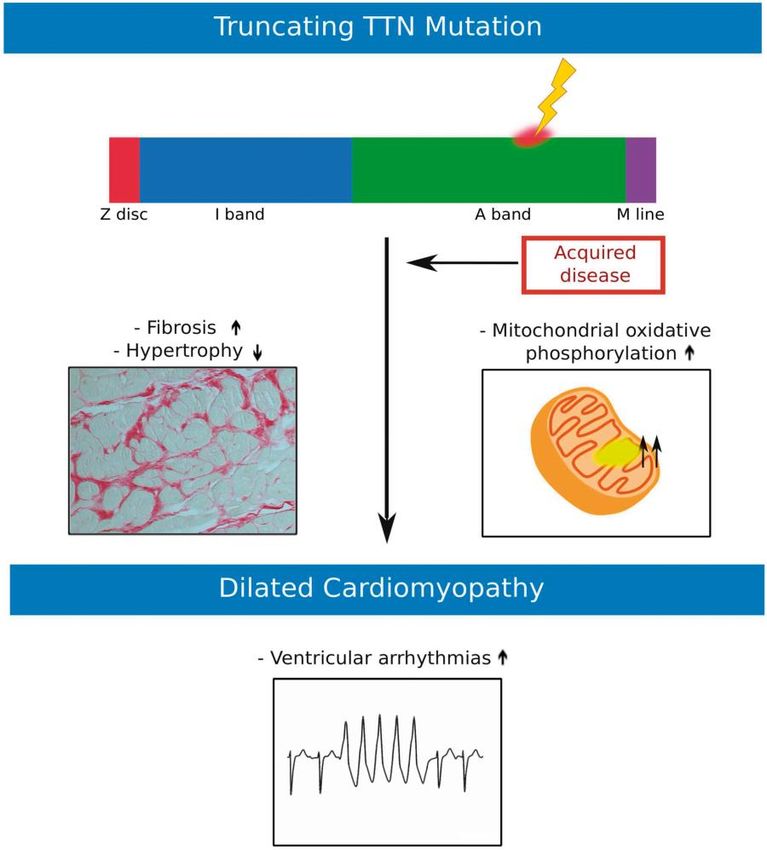
USB Sprechstunde 3.1.20 12-Kanal EKG
Kardiologie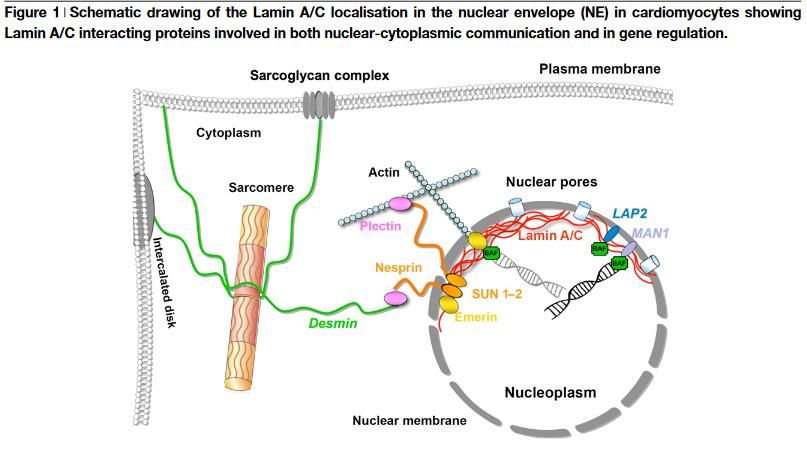
Rhythmusstreifen und Holter
Kardiologie
Sprechstunde USB 3.1.2020 41 jährige Akademikerin (2 Kinder) kommt mit Gattin und Schwägerin zur Besprechung eines DDD-SM, ggf. His-Bundle Pacing Seit 10 Jahren VES (VES Last 7%), keine Synkopen oder Präsynkopen Ausgeprägter AV-Block I im Wechsel mit AV-Block II Typ Wenckebach Patientin beginnt zu weinen und sagt, sie habe Angst das gleiche Schicksal wie Ihre Mutter zu haben, die 1997 48 jährig mit der Diagnose einer dilatativen CMP auf der HTx Liste verstarb
Prozedere Sprechstunde
Anbindung Psychosomatik
KardiologieLamin A/C Cardiomyopathie
Lamin A ist ein Strukturprotein
des Zellkernmembran, welches
filamentär die Kernmembran und
die Kernmatrix trennt.
LMNA Gen kodiert für die beiden
Isoformen Lamin A und C
Laminopathien betreffen Herz-
und Skelettmuskel
Hutchinson-Gilford Progerie
Syndrom («Voraltern» und Tod
wegen MI oder Stroke im
Teenageralter)
Carmosino M et al., Biol Cell 2014;106:346
Kardiologie«Familiäre dilatative Cardiomyopathie»
In 20-50% der DCM-Fälle kann eine Mutation in einem von 50 Genen
nachgewiesen werden
KardiologiePhänotypisierung und Genetik
Chen SN et al., Cur Cardiol Rep 2019;21:160
KardiologieMolekulargenetische Analyse
KardiologieWie weiter?
I. «Wait and see», da normale LVEF und keine Synkopen
II. Herzinsuffizienztherapie
III. DDD-Schrittmacherimplantation (His-bundle pacing?)
IV. DDD-ICD Implantation
V. Implantierbarer Loop-Rekorder
KardiologieLamin A/C Cardiomyopathie
Single Center Studie aus Oslo
Zwischen 2003-2015 wurden 561 Patienten genetisch für familiäre DCM
getestet -> 6.2% mit LMNA Mutation (18 verschiedene Mutationen)
Hasselberg NE at al, Eur Heart J 2018;39:853-860
KardiologieLamin A/C Cardiomyopathie - Outcome
Hasselberg NE at al, Eur Heart J 2018;39:853-860
Kardiologiewww.lmna-risk-vta.fr
KardiologieTherapie
DDD-ICD Implantation
Beginn mit ACE-Hemmer Therapie
(LVEDD 56 mm)
Vorstellung neuromuskuläre Sprechstunde:
Prox. betonte Muskelschäche
untere Extremität
Chen SN et al., Cur Cardiol Rep 2019;21:160
KardiologieAetiologie Dilatative Cardiomyopathien
KardiologieAetiologie dilatative Cardiomyopathie
DCM
Div. 65-80% 20-35%
Familiär
Aetiologien
• Strukturell
• Kongenital
• Drogen/Medi
• Endokrin
• Immun-mediiert
• Infiltrativ
• Infektiös
• Metabolisch
• Toxine
• Strahlung
• Tachykardie
KardiologieKardiologie Von Eugène Delacroix - Erich Lessing Culture and Fine Arts Archives via artsy.net, Gemeinfrei, https://commons.wikimedia.org/w/index.php?curid=27539198
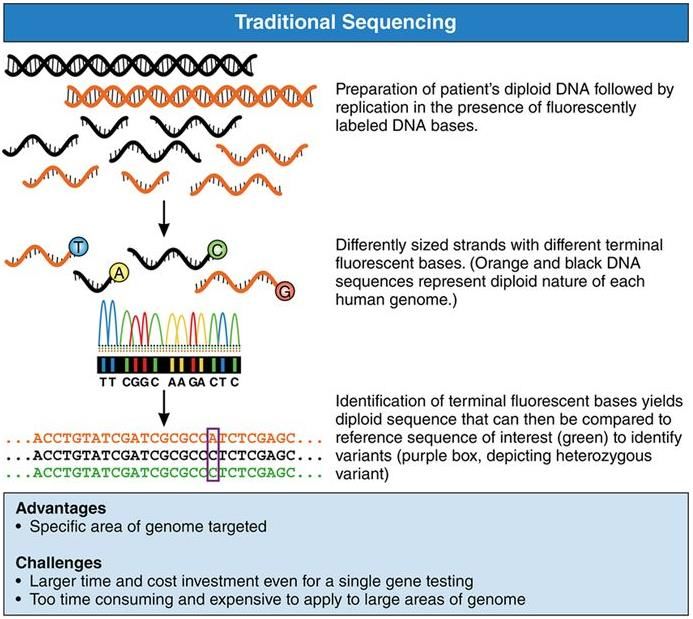
Didesoxy-Sequenzierung nach Sanger
Kardiologie
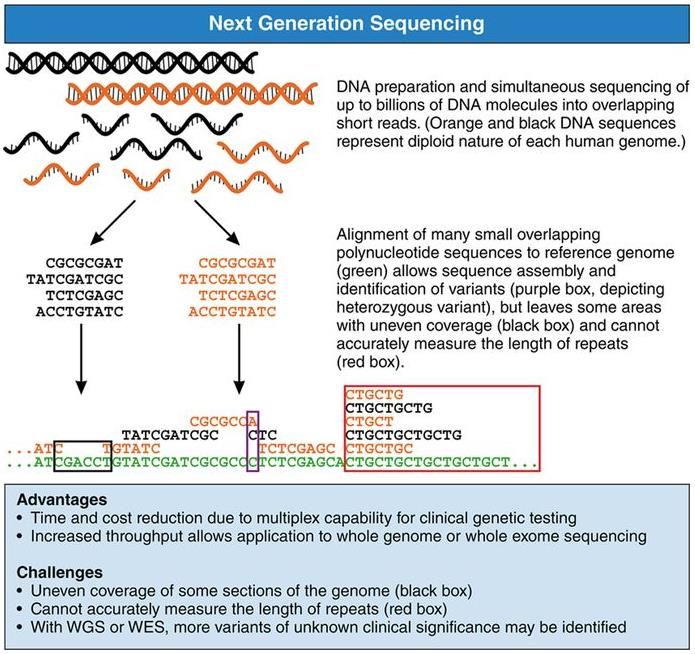
Parikh VN et al., Circulation 2017;406-409Next generation sequencing
Kardiologie
Parikh VN et al., Circulation 2017;406-409Aetiologische Charakterisierung DCM
Kardiologie
Merlo M et al., Eur J Heart Fail 2018;228-239Bsp. Aethyltoxische Kardiomyopathie
KardiologieVarianten bei familiärer dilatative Cardiomyopathie
KardiologieKardiomyozyt
Kardiologie
Paladino A et al., Curr Cardiol Rep 2018Genotyp-Phänotyp-Korrelationen
Kardiologie
Merlo M et al., Eur J Heart Fail 2018;228-239Kardiologie Kayvanpour E et al., Clin Res Cardiol 2017;127-139
Titin
Kardiologie
Verdonschot JAJ et al., Eur Heart J 2018;864-873Empfehlung ESC zur genetischen Testung
«Genetische Testung ist bei familiären DCM-
Formen ODER bei sporadischer DCM mit
zusätzlichen klinischen Hinweisen welche eine
genetische Ursache suggerieren empfohlen.»
Kardiologie
Pinto YM et al., Eur Heart J 2016;1850-8Evaluation genetischer Testung: 1. Schritt
Kardiologie
Favalli V et al., Heart 2016;2004-2014Klinische Hinweise für genetische Ursachen
Pinto YM et al., Eur Heart J 2016;1850-8
Gorbatsch S et al., J Dtsch Dermatol Ges 2020;50-54
Kardiologie
Srinivas SM et al., Int J Trichology 2016;53-55EKG-Hinweise für genetische Ursachen
Pinto YM et al., Eur Heart J 2016;1850-8
Buckley AE et al., Heart 1999, 105-108 Kardiologie
Captur G et al., Heart 2018, 468-479Hinweise im Labor für genetische Ursachen
Kardiologie
Pinto YM et al., Eur Heart J 2016;1850-8Abklärungs-Algorithmus
• Reizleitungsstörung
• Relevante ventrikuläre
Arrhythmien
• Atypische atriale
Arrhythmien
• Skelettmuskelbeteiligung
• EKG-Auffälligkeiten
• TTE (posterolat. Akinesie)
Kardiologie
Paladino A et al., Curr Cardiol Rep 2018Praktisches Vorgehen
Patient muss informiert und beraten werden!
Schriftliche Einwilligung
Kostengutsprache anfordern
Patient kann hierfür der HALB-Sprechstunde zugewiesen werden
KardiologieHALB-Sprechstunde
Hypertrophe, Arrhythmogene Kardiomyopathien, Long-QT, Brugada
Patienten werden GANZ beurteilt
Inhalt der Sprechstunde
Diagnosestellung
Therapie inkl. Indikationsstellung für interventionelle Eingriffe
Beratungen z.B. bzgl. Sport
Genetische Abklärungen/Familienberatung
Zuweisung: E-Mail an annina.vischer@usb.ch, übliche Kanäle für
Zuweisung Kardiologie oder Medizinische Poliklinik
«Rare things are only rare if you don’t look for them»
KardiologieTake Home Messages
«Dilatative Kardiomyopathie» ist eine morphologischen Beschreibung
Ursachen äusserst heterogen – entsprechend auch die Prognosen
Rund ein Drittel der DCM-Fälle sind familiär
Genetische Ursache sind aber auch in nicht familiären Formen häufig
Eine genetische Abklärung kann in gewissen Fällen zur Risikostratifizierung
beitragen
Kardiologiewww.cardiobasel.ch
Sie können auch lesen

















































