Molekulargenetische Untersuchungen zur Populationsdifferenzierung von Rattus rattus L. auf Madagaskar - Melanie Münster
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

Molekulargenetische Untersuchungen zur
Populationsdifferenzierung von
Rattus rattus L. auf Madagaskar
von
Melanie Münster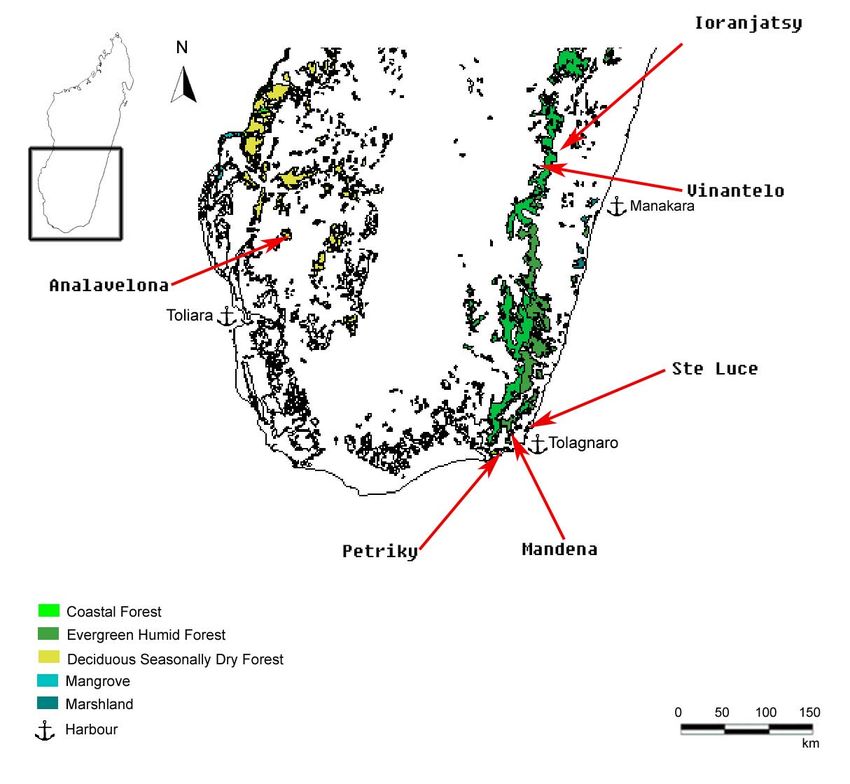
Molekulargenetische Untersuchungen zur
Populationsdifferenzierung von
Rattus rattus L. auf Madagaskar
Diplomarbeit
am Fachbereich Biologie
der Universität Hamburg
Erstgutachter: Prof. Dr. Jörg U. Ganzhorn
Zweitgutachter: Prof. Dr. J. Parzefall
vorgelegt von
Melanie Münster
Hamburg, September 2003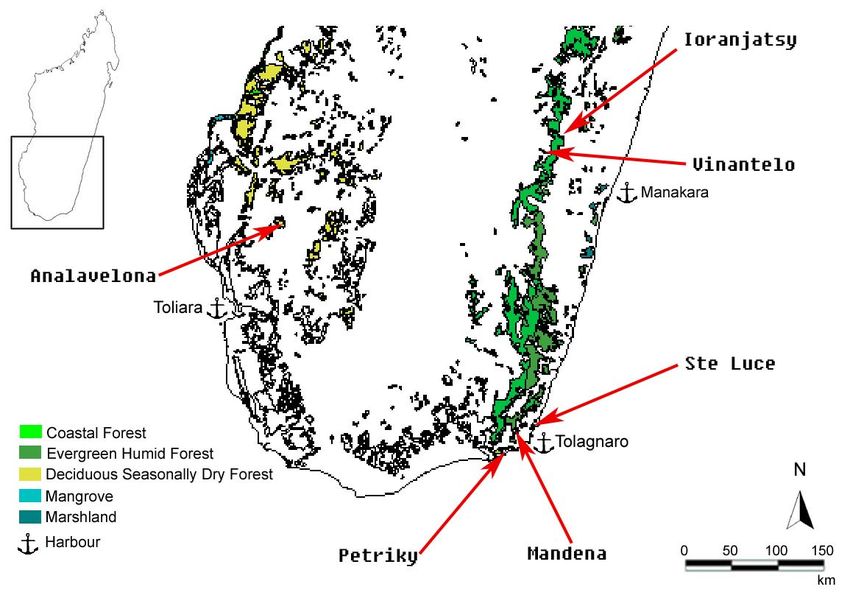
Ny hazo tokano tsy mba ala One tree doesn´t make a forest

Inhaltsverzeichnis
Inhaltsverzeichnis
1. Einleitung....................................................................................................... 1
2. Material und Methoden ................................................................................. 6
2.1 Untersuchte Art Rattus rattus................................................................... 6
2.2 Untersuchungsgebiete und Probenmaterial ............................................. 8
2.2.1 Gebietsbeschreibungen .................................................................... 8
2.2.2 Probenmaterial................................................................................ 13
2.3 Molekulargenetische Methoden ............................................................. 14
2.3.1 DNA Extraktion ............................................................................... 14
2.3.2 Agarose-Gelelektrophorese ............................................................ 15
2.3.3 PCR - Polymerase Chain Reaction................................................. 16
2.3.4 Ethanolfällung ................................................................................. 19
2.3.5 Single Strand Conformation Polymorphism (SSCP) ....................... 19
2.3.6 Sequenzierung................................................................................ 22
2.4 Auswertungsmethoden .......................................................................... 23
3 Ergebnisse................................................................................................... 27
3.1 mtDNA ................................................................................................... 27
3.1.1 Zielfragment Hypervariable Region I (HVR I).................................. 27
3.1.2 Variabilitätsanalyse der HVR I ........................................................ 28
3.1.3 Genetische Variabilität innerhalb der Populationen ........................ 33
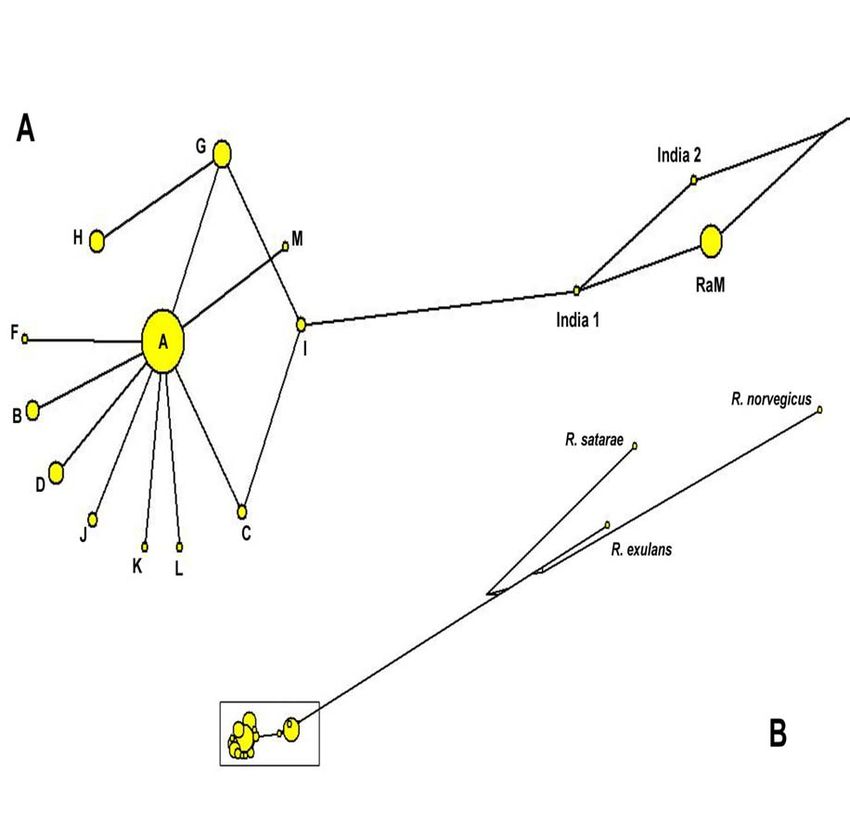
3.1.4 Populationsdifferenzierung ............................................................. 35
3.1.5 Potentielle Invasionsherde.............................................................. 37
3.1.6 Selektive Neutralität ........................................................................ 38
3.1.7 Biogeographie................................................................................. 40
3.2 MHC....................................................................................................... 43
3.2.1 Zielfragment Exon 2 des MHC DRB Genes .................................... 43
3.2.2 Variabilität des DRB Exon 2 Genortes ............................................ 44
3.2.3 Selektionsmechanismen in den Antigenbindungsstellen ................ 46
3.3 Variabilitätsvergleich der Marker ............................................................ 47
4. Diskussion ................................................................................................... 49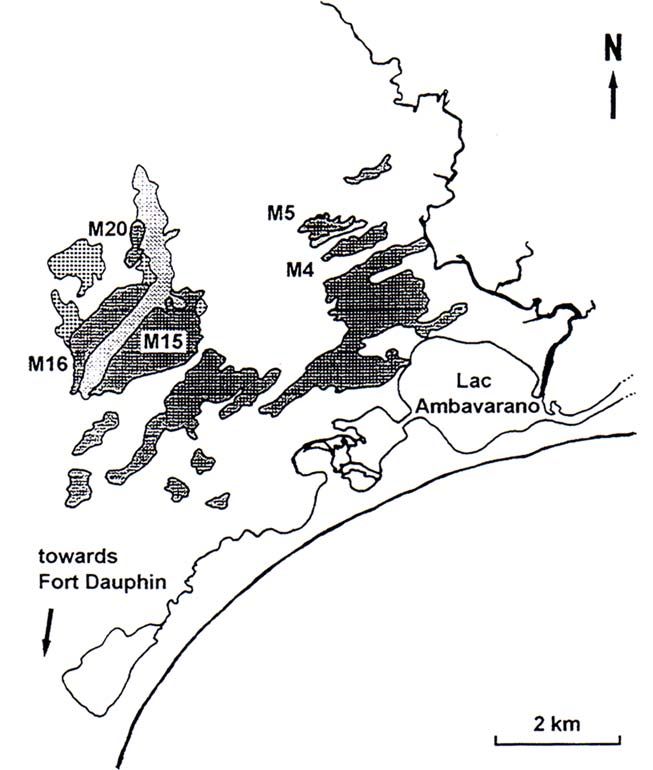
Inhaltsverzeichnis
5. Zusammenfassung ..................................................................................... 59
6. Literatur........................................................................................................ 61
7. Abkürzungen ............................................................................................... 76
ANHANG I - Labormaterialien............................................................................... I
A Chemikalien ....................................................................................... I
B Kits..................................................................................................... I
C Verbrauchsmaterial........................................................................... II
D Lösungen .......................................................................................... II
E Laborgeräte...................................................................................... IV
ANHANG II - Probenübersicht ............................................................................. V
ANHANG III - mtDNA Alignment......................................................................... IX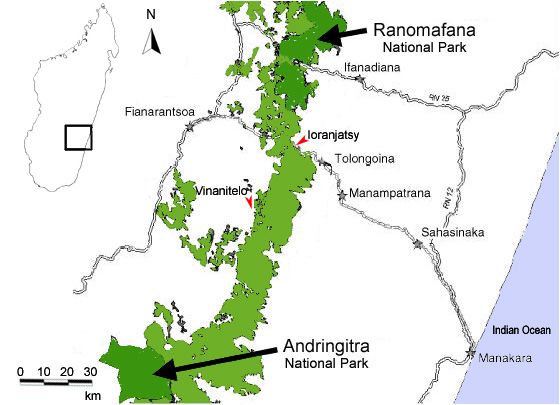
1. Einleitung 1 1. Einleitung Invasive Arten Der Einfluss invasiver Arten auf einheimische Arten, ihre Gemeinschaften und Ökosysteme ist bereits seit einigen Jahrzehnten bekannt (PARKER et al. 1999; ELTON 2000; SAKAI et al. 2001). Er wird heute als wesentlicher Bestandteil der globalen Veränderung und, nach dem Verlust und der Degradierung von Habitaten, weltweit als grösste Bedrohung für die Biodiversität angesehen (VITOUSEK et al. 1996; WILCOVE et al. 1998; WILSON 1999; FRANKHAM et al. 2002). Neben absichtlich als Haus- oder Nutztier eingeführten Arten wurden seit dem 15. Jahrhundert im Zuge der Erforschung (Kolonialisierung) der Kontinente durch die Europäer, und werden heute durch den weltweiten Handel, Pflanzen und Tiere als „blinde Passagiere“ über lange Distanzen verbreitet (O.T.A. 1993; MCNEELY 2001). Nicht jede eingeschleppte Art ist jedoch auch eine invasive Art. Essentiell für die Bezeichnung „invasiv“ ist das Maß ihrer Auswirkungen auf die neue Umwelt (DAVIS & THOMPSON 2000). Diese Auswirkungen beinhalten zum einen den Effekt der Konkurrenz, indem eine invasive Art die verfügbaren Ressourcen für andere Arten reduziert und zum anderen den Effekt auf das Ökosystem, indem sie die elementaren Gegebenheiten des Ökosystems modifiziert (DUKES & MOONEY 1999). Mindestens drei Stadien gehören zu einer erfolgreichen Invasion: nach (1) der Einführung in ein neues Gebiet muss es (2) zu einer sich erfolgreich etablierenden Erstkolonialisierung kommen, von der aus (3) eine nachfolgende Verbreitung in neue Habitate ausgeht (SAKAI et al. 2001). Der „enemy release Hypothese“ nach haben invasive Arten den einheimischen Konkurrenten gegenüber viele Vorteile (KEANE & CRAWLEY 2002). Sie haben primär keine natürlichen Feinde und ihre Belastung durch Parasiten und Krankheiten ist geringer als in ihrem ursprüglichen Habitat (TORCHIN et al. 2001; CLAY 2003; TORCHIN et al. 2003). Der Begriff „invasional meltdown“ wurde eingeführt um die Akkumulation von biologischen Invasionen zu beschreiben, die in ihrem Erfolg aufeinander aufbauen. Ein Beispiel hierfür bieten die Habitatveränderungen durch den Menschen (Homo sapiens sapiens), welche verschiedenen invasiven Arten erst den nötigen Vorteil verschafft, sich durchzusetzen (SIMBERLOFF & VON HOLLE 1999). Die Etablierung in einer natürlichen Artengemeinschaft benötigt andere Charakteristika, als das Eindringen in ein anthropogen gestörtes Habitat (HORVITZ et al. 1998). Studien der Populationsbiologie von invasiven Arten ermöglichen einen genaueren Einblick in die Charakteristika, die ausschlaggebend sind für ihre Invasivität (CRAWLEY 1986). Die meisten invasiven Wirbeltiere sind mit dem Menschen eng verbunden (Koevolution) und haben wie wir ein breites
1. Einleitung 2 Nahrungsspektrum (EHRLICH 1989). Auch tolerieren viele Vertebraten eine grössere Spannbreite physischer Bedingungen als sie in ihrem natürlichen Lebensraum begegnen (BROWN 1989). Generell ist Toleranz für eine heterogene Umwelt (Phenotypische Plastizität) von Vorteil (SAKAI et al. 2001), ebenso eine hohe Reproduktionsrate und die Konkurrenzfähigkeit einer invasiven Art (MACARTHUR & WILSON 1967; PIANKA 1970). Viele Invasionen führen erst nach einer langen Latenzzeit oder nach der mehrfachen Einführung einer Art zum Erfolg, und die Invasoren, die innerhalb historischer Zeit erfolgreich eine neue Region besiedelt haben, sind lebende Beispiele für einen ökologischen und evolutionären Wandel (ELLSTRAND & SCHIERENBECK 2000). Diese Invasionen können auch als natürliche Experimente gesehen werden, da populationsökologische Prozesse möglicherweise viel schneller ablaufen als in einem ungestörtem System (SAKAI et al. 2001). Da eine Kolonialisierung meist von wenigen Individuen einer Ursprungspopulation ausgeht, ist die neue Population durch diesen Flaschenhalseffekt genetisch nicht so differenziert wie die, von der sie abstammt (TSUTSUI et al. 2000). Madagaskar Madagaskar ist die viertgrösste Insel der Erde. Ihre Fläche entspricht mit 587.000 km2 etwa 0,4% der terrestrischen Landoberfläche. Seit 165 Millionen Jahren von anderen Landmassen getrennt, liegt sie heute etwa 350 km östlich vor dem Afrikanischen Kontinent (LOWRY II et al. 1997; GARBUTT 1999; TYSON 2000). Durch diese Isolation konnte sich auf Madagaskar eine einmalige Flora und Fauna entwickeln, deren Endemismenrate jeweils etwa 80% erreicht und sich über das Artniveau hinaus bis zu Gattungen und Familen erstreckt (DREIER 1992; MYERS et al. 2000; TYSON 2000; WWF 2001). Die ursprüngliche Vegetation hat seit der Besiedlung durch den Menschen vor etwa 2000 Jahren stetig abgenommen. Diese Verluste, die hauptsächlich durch Brandrodung zustandegekommen sind, haben dazu geführt, dass heute nur noch etwa 9,9% der Primärvegetation existieren (RABINOWITZ et al. 1983; DEWAR 1997; MACPHEE & MARX 1997; MYERS et al. 2000). Allein in den Jahren zwischen 1950 und 1983 sind jährlich 111.000 ha entwaldet worden (GREEN & SUSSMAN 1990). Die dadurch verursachte Habitatzerstörung und -fragmentierung stellt für viele einheimische Arten ein kaum überwindbares Hindernis dar. Nachfolgende Sekundärvegetation besteht neben dem kleinen Anteil an Farmland, Eukalyptusforsten und Kiefernwäldern hauptsächlich aus sekundärem Grassland (formation herbeuse) oder verholztem Grassland (formation herbeuse boisée), die nur durch ihre Artenarmut herausstechen (WHITE 1983; LOWRY II et al. 1997).
1. Einleitung 3 Madagaskar gehört weltweit zu den drei am meisten gefährdeten Hotspots für Biodiversität. Dazu tragen die hohe Endemismenrate, die Artenvielfalt bezogen auf die Fläche und ihr Bedrohungszustand bei. Nur 20% der verbliebenen Primärwaldfläche werden bereits geschützt (MYERS et al. 2000; TYSON 2000). Der mit der Habitatzerstörung einhergehende Biodiversitätsverlust erleichtert zusätzlich die Invasion durch eingeführte Arten (STACHOWICZ et al. 1999; WESTERN 2001; KENNEDY et al. 2002) und deren Verbreitung bedroht durch Konkurrenz oder Prädation die endemische Fauna Madagaskars (RAMANAMANJATO & GANZHORN 2001). Die häufigsten fremden Tierarten sind die menschlichen Kommensalen Ratte (Rattus rattus) und Maus (Mus musculus), die an die durch den menschlichen Einfluss entstehenden Lebensraumveränderungen seit Generationen angepasst sind (LOWRY II et al. 1997; GARBUTT 1999). Die Ratte steht im Verdacht die auf Madagaskar endemischen Nesomyinae hauptsächlich durch Nahrungskonkurrenz zu verdrängen (GOODMAN 1995), wobei ein negativer Einfluss von R. rattus auf die einheimische Nagetierfauna zur Zeit jedoch nicht bestätigen werden konnte (GANZHORN 2003). Informationen über die Genetik, Evolution und Interaktionen von invasiven und einheimischen Arten in betroffenen Gemeinschaften können Aufschlüsse über die Anfälligkeit eines Ökosystems für Invasionen geben (SAKAI et al. 2001). Molekulare Marker Besonders die phylogenetische Analyse von DNA oder Proteinsequenzen ist ein gutes Instrument für Studien der Molekularen Evolution geworden (NEI & KUMAR 2000). Für die Beantwortung vieler populationsgenetischer Fragestellungen sind Marker, die keinem Selektionsdruck unterliegen, von Vorteil (SUNNUCKS 2000). Die DNA der nichtkodierenden Kontrollregion der Mitochondrien ist ein solcher neutraler Marker. Mitochondrien sind in der Zelle vorwiegend für die oxidative Phosphorilierung und die Bildung von ATP zuständig (CASTRO et al. 1998). Es handelt sich bei ihnen um Relikte endosymbiontischer (Purpur-) Bakterien, die mit Zellen eine stabile Beziehung eingingen (MADIGAN et al. 2001). Aufgrund dieser Herkunft verfügen sie über eine eigene haploide DNA, die üblicherweise nicht rekombinant ist, sich autonom vermehrt (CASTRO et al. 1998) und bis auf wenige Ausnahmen ausschliesslich maternal vererbt wird (AVISE et al. 1987; EYRE- WALKER et al. 1999; WALLIS 1999; BROMHAM et al. 2003). Die mtDNA ist bis auf wenige Ausnahmen kovalent ringförmig geschlossen und umfasst etwa 16.500 bp. Die kodierenden Gene der Mitochondrien sind konservativ und umfassen zwei ribosomale (rRNA) Gene, je eines für die 12S und 16S rRNA, 22 transfer RNA (tRNA) Gene und 13 proteinkodierende Gene (Anderson et al. 1981). Für populationsgenetische Fragestellungen ist die Kontrollregion, der sogenannte
1. Einleitung 4 ’displacement loop’ (d-loop) von besonderem Interesse (RICHARDS & MACAULAY 2001). Die Kontrollregion ist bekannt für ihre hohe Mutationsrate, sowohl für Nukleotid-Substitutionen als auch für Insertionen/ Deletionen (BROWN et al. 1979; BROWN & SIMPSON 1982; MORITZ et al. 1987). Bei Menschen und Nagern ist ausserdem eine höhere Rate von Transitionen als Transversionen nachgewiesen worden (AQUADRO & GREENBERG 1983; BROWN et al. 1986). Anhand von Studien an der menschlichen mtDNA wurde der d-loop in drei Bereiche eingeteilt, zwei hypervariable Regionen (HVR I & HVR II) und einen dazwischen liegenden konservativen Block (WALBERG & CLAYTON 1981; VIGILANT et al. 1991; MATSON & BAKER 2001). Die Mutationsrate innerhalb der hypervariablen Regionen ist etwa zehnmal so gross wie die der proteinkodierenden Region der mtDNA (VIGILANT et al. 1991; PESOLE et al. 1999; RICHARDS & MACAULAY 2001). Wegen der aufgezählten Charakteristika der mitochondriellen DNA ist sie und besonders die HVR I des d-loops zu einem beliebten Marker der Evolutionsbiologie geworden. Mit ihrer Hilfe lassen sich phylogenetisch Beziehungen zwischen eng verwandten Arten und zwischen Populationen innerhalb einer Art auflösen (AVISE 1986; MORITZ et al. 1987; HARRISON 1989). Aufgrund ihrer funktionellen Wichtigkeit gewinnen die kodierenden Immungene des ’Major Histocompatibility Komplex’ (MHC) für vergleichende Untersuchungen zur genetischen Diversität von Tierarten und der potentiellen Konsequenzen bei Verlust genetischer Diversität zunehmend an Bedeutung (VAN DER WALT et al. 2001; SOMMER 2003). Der MHC ist eine Genfamilie, die entsprechend ihrer Funktion und Struktur in drei Klassen eingeteilt wird. Die Expression der MHC Klasse I kodierten Moleküle, findet auf allen Zelloberfächen ausser auf der der Erythrozyten statt. Diese MHC-Proteine binden Peptide im Cytosol und sind für die Unterscheidung von selbst und nicht selbst z. B. bei Gewebetransplantationen verantwortlich und daher namensgebend für den Komplex (Gewebeverträglichkeit – histocompatibility) (GRUEN & WEISSMAN 1997). Drei Genloci (beim Menschen A, B, C) kodieren für je eine Kette. Die Expression der MHC Klasse II Moleküle findet nur auf den antigenpräsentierenden Zellen statt (B-Lymphozyten, Makrophagen). Sie sind für die zellvermittelte Immunantwort im interzellulärem Raum (niedriger pH) verantwortlich, wie sie z. B. gegenüber Bakterien eingesetzt wird. Ebenfalls drei Genloci sind bekannt, DR, DQ und DA, die ihrerseits für eine α und eine β Kette kodieren. Die Klasse III kodierten Moleküle sind am Komplementsystem beteiligte Proteine, die frei im Serum vorliegen. Sie sind an der humoralen Immunantwort beteiligt. Zwischen einigen Genloci, vor allem zwischen DR und DQ und zwischen B und C liegt ein ’Linkage Disequilibrium’ (Koppelungs Ungleichgewicht) vor. Dies bedeutet, dass eng zusammenliegende Gene
1. Einleitung 5 gemeinsam vererbt werden. Die Gene des MHC sind sehr polymorph und gehören zu den variabelsten Genen, die bei Vertebraten bekannt sind (HEDRICK 1994). Diese Diversität wird durch Selektionsprozesse aufrecht erhalten (BERNATCHEZ & LANDRY 2003). Die möglichen Selektionsmechanismen basieren einerseits auf pathogen- bzw. parasitengesteuerter Selektion und andererseits auf Mechanismen, die durch Reproduktionsvor- bzw. nachteile gelenkt werden. Die Theorie des Heterozygotenvorteils (’overdominant selection’) geht davon aus, dass durch balancierende Selektion ein breites Abwehrspektrum gegen koevolvierende Pathogene und Parasiten aufrecht erhalten wird. Die Theorie des ’rare Allele Advantage’ (oder auch ’Red-Queen’-Hypothese) hingegen sieht den Vorteil einzelner Allele, für den Fall, das Parasiten oder Pathogene durch eigene Evolution die allgemein verbreitete Immunabwehr umgangen haben (zusammengefasst in PENN & POTTS (1999) und BERNATCHEZ & LANDRY (2003)). In diesem Zusammenhang steht auch die ’moving target’-Hypothese von PENN & POTTS (1999), die wiederum ein koevolutives ’Wettrüsten’ mit Parasiten beschreibt. Reproduktionsbasiert wird zum einen die Theorie der ’inbreeding avoidance’ (POTTS & WAKELAND 1990; 1993) diskutiert, die von MHC-abhängigen Paarungspräferenzen (disassortative mating) z. B. anhand von Gerüchen ausgeht, um Inzucht zu vermeiden. Zum anderen ist auch die erhöhte Fitness der Nachkommen selektiv bevorteilter Individuen durch ’disassortative mating’ möglich. Eine balancierende Selektion auf dem molekularen Level dieser Loci manifestiert sich durch ein erhöhtes Verhältnis von nichtsynonymer zu synonymer Substitution (dN/dS) an den funktionell wichtigen Antigenbindungsstellen (ABS) (HUGHES & NEI 1988; YANG & NIELSEN 2002). Nach BROWN et al. (1993) sind 27 Aminosäuren der β1-Domäne der MHC Klasse II Proteine mutmasslich an der Antigenbindung beteiligt. Ziel dieser Arbeit war es, am Beispiel der auf Madagaskar eingeschleppten Art Rattus rattus zu zeigen, wie hoch die Variabilität eines selektionsneutralen Markers (HVR I des mitochondriellen d-loops) dieser invasiven Art innerhalb der Inselpopulation Madagaskars ist. Auch sollte untersucht werden, ob die Variabilität des Markers innerhalb verschiedener Regionen Madagaskars unterschiedlich ausgeprägt ist und mögliche Invasionsherde festgestellt werden. Ein weiteres Ziel war, durch Vergleich mit R. rattus-Proben aus anderen Orten der Welt das Herkunftsgebiet der madagassischen Ratten zu klären. Zusätzlich sollte untersucht werden, wie sich die Variabilität des nichtkodierenden Bereiches (HVR I des d-loops der mtDNA) zu der Variabilität eines kodierenden Genortes des Immunkomplexes (MHC DRB Exon 2) verhält.
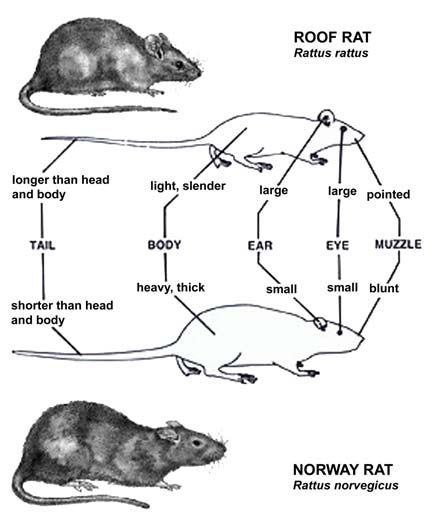
2. Material und Methoden 6 2. Material und Methoden 2.1 Untersuchte Art Rattus rattus Die Hausratte Rattus rattus (LINNAEUS, 1758) gehört innerhalb der grössten Säugetierordnung, den Rodentia, zu der Familie der Muridae, den echten Mäusen. Ursprünglich kam sie in den warmen Regionen Südostasiens, wahrscheinlich in Indien vor und wurde von dort aus mit Schiffen in alle Teile der Welt verschleppt (STORCH & WELSCH 1997; NOWAK 1999). Es sind mittelgrosse, bis ca. 200 g schwere, schwarze bis schwarzbraune schlanke Tiere, deren Schwanz meist länger als Kopf und Rumpf ist (Abb. 2) (DAVIS & SCHMIDLY 1994). Die ursprünglichen Baumbewohner sind dämmerungs- und nachtaktiv, exzellente Kletterer (Abb. 1) und bauen in geeigneten Schlupfwinkeln, wie etwa in Bäumen oder anderen Erhöhungen, kugelförmige Nester aus verschiedensten verfügbaren Materialien (KENNEDY 1993; NOWAK 1999). Abb. 1: Rattus rattus in seinem ursprünglichen Habitat, Photo von Robert L. Curry, Hawaii. Sie leben einzeln oder in Gruppen bis zu 50 Tieren (BECKER 1978; STORCH & WELSCH 1997). Zwei bis drei gleichrangige Weibchen sind einem dominanten Männchen untergeordnet, jedoch den weiteren Gruppenmitgliedern übergeordnet (EWER 1971; GARBUTT 1999). Feste Rangordnungen oder Paarbindungen lassen sich nicht nachweisen (MOHR 1954; TELLE 1966). Die allgemeine Aggressionsbereitschaft ist sehr gering (♀ > ♂) und beschränkt sich auf die Verteidigung des rund um den Futterplatz gelegenen Gruppenterritoriums gegen
2. Material und Methoden 7
Eindringlinge (NOWAK 1999). Die Fortpflanzung erfolgt meist ganzjährig. Das
bereits mit 3 Monaten geschlechtsreife Weibchen bringt nach 21-23 Tagen
Tragzeit durchschnittlich 8 Junge zur Welt (DAVIS & SCHMIDLY 1994; KAESTNER &
STARCK 1995). Hausratten ernähren sich bevorzugt von Samen, Körnern, Nüssen,
Früchten und Gemüse, aber auch Insekten und andere Invertebraten finden sich
in ihrem Futterspektrum (BECKER 1978; GARBUTT 1999; NOWAK 1999).
R. rattus gehört zu den sieben
bedeutendsten Rattenarten, die sich
als menschliche Kommensalen
durchgesetzt haben und in ihrer
Verbreitung direkt von
Transportwegen oder indirekt durch
Habitatveränderungen profitieren
(NOWAK 1999). Sie ist wahrscheinlich
das erste Säugetier, welches sich mit
den frühen Menschen assoziiert hat.
Ihre Ausbreitung von Indien nach
Ägypten erfolgte bereits im 4ten
Jahrhundert BC und daraufhin über alle
Handelswege auch nach Europa
(TAYLOR 1964). Seinen englischen Abb. 2: Vergleichende Darstellung von
Namen ’black rat’ hat R. rattus Schiffsratte (Rattus rattus, oben) und
erhalten, da die Art mit dem Wanderratte (Rattus norvegicus). Unterschiede
’schwarzen Tod’ (black death) in sind ersichtlich anhand der Ohren, der
Schnauze und des Schwanzes.
Verbindung gebracht wird.
Nachgewiesen ist, dass sie dem Rattenfloh Xenopsylla cheopis als Wirt dient, der
als Vektor des Bubonenpesterregers Yersinia pestis bekannt ist (MADIGAN et al.
2001). Mit Beginn des 18ten Jahrhunderts wurde die sogenannte Schiffsratte
innerhalb Europas (temperierte Gebiete) graduierlich von der Wanderratte (Rattus
norvegicus) verdrängt. Generell ist die wärmeliebende Art heute in urbanen
Regionen vornehmlich in tropischen Klimaten anzutreffen (NOWAK 1999). R. rattus
ist als invasive Art besonders auf Inseln sehr erfolgreich und richtet durch ihre
Fähigkeit sowohl arboreal als auch auf dem Grund leben zu können, massive
Schäden an der einheimischen Fauna an (nach KING 1985 in AMORI & CLOUT
(2003)). Auf Madagaskar wurde R. rattus wahrscheinlich bereits im 11ten
Jahrhundert eingeführt. Die Art hat sich möglicherweise aber erst seit der
Jahrhundertwende 1900 bis in die entlegendsten Waldgebiete verbreitet
(GOODMAN 1995; GARBUTT 1999).2. Material und Methoden 8 2.2 Untersuchungsgebiete und Probenmaterial Die in dieser Arbeit untersuchten Proben stammen aus insgesamt sechs Untersuchungsgebieten (Abb. 3) der Südhälfte Madagaskars. Die Probenorte können zu drei übergeordneten Gebieten zusammengefasst werden, der Province de Toliara, der Province de Fianarantsoa und der Tolagnaro Region (Fort Dauphin-Region). Sie unterscheiden sich in Lage, Vegetation und Klima. 2.2.1 Gebietsbeschreibungen Westen - Province de Toliara: Der Südwesten Madagaskars, zu dem auch die Province de Toliara mit ihrem gleichnamigen Überseehafen gehört, hat mit 360–660 mm pro Jahr die geringsten Niederschlagsmengen der Insel (DONQUE 1972). Neben einer durchschnittlichen Jahrestemperatur von 30°C dauert die Trockenzeit 6 bis 8 Monate an. Daher wird die natürliche Vegetation von laubabwerfenden Bäumen und von stark spezialisierten Pflanzen wie z. B. Euphorbiaceen beherrscht (KOECHLIN 1972). Abb. 3: Die Lage der Probengebiete Analavelona, Ioranjatsy, Vinantelo, Ste Luce, Mandena und Petriky (rote Pfeile) in Südmadagaskar. Farbig dargestellt ist die erhaltene Primärvegetation, die Anker verweisen auf nahe Überseehäfen. Die Darstellung basiert auf GIS-Daten von DU PUY & MOAT (1996).
2. Material und Methoden 9 Forêt d'Analavelona In der Province de Toliara liegt das natürlich isolierte Feuchtwaldplateau von Mount Analavelona (Abb. 3). Der verbliebende Regenwald auf dem ca. 1250 m über NN liegenden Plateau ist von degradiertem Trockenwald, grossen Grasslandgebieten und exotischen Baumplantagen umgeben, die das Produkt menschlicher Aktivitäten sind. Die lokale Rattenpopulation ist sehr gross. Es gibt keine Hinweise auf Beulenpestvorkommen (GOODMAN, mündliche Mitteilung). Osten - Province de Fianarantsoa: Der Ranomafana-Andringitra-Korridor (Abb. 4) in der Province de Fianarantsoa ist ein etwa 160 km langer, 4-15 km breiter Abschnitt des verbliebenen Küstenregenwaldstreifens der Ostküste, welcher den Andringitra National Park mit dem Ranomafana National Park verbindet. Die Niederschläge nehmen von der Küste im Osten (2500 mm / Jahr) zum Hochplateau im Westen (1500 mm / Jahr) ab (nach HUMBERT & COURS DARNE 1965 in KOECHLIN (1972). Aufgrund der grossen Höhendifferenzen auf begrenztem Raum ist das Ökosystem wegen seiner Biodiversität besonders interessant. Das Waldgebiet wird jedoch insbesondere in geringeren Höhen (500–800 m) durch die sogenannte Tavy Landwirtschaft, die auf Urbarmachung durch Abholzung und Brandrodung basiert, zunehmend fragmentiert (FREUDENBERGER & FREUDENBERGER 2002). Der nächstgelegene Überseehafen liegt südöstlich in Manakara. Abb. 4: Der Ranomafana-Andringitra-Korridor mit den Probenstellen Ioranjatsy und Forêt de Vinantelo (verändert nach FREUDENBERGER & FREUDENBERGER (2002)).
2. Material und Methoden 10 Ioranjatsy Ioranjatsy ist ein kleines Dorf am Rande des Regenwaldes des Ranomafana- Andringitra-Korridors (Abb. 3 & Abb. 4). Gefangen wurde hauptsächlich im dortigen Tavy-Wald-Gebiet nahe der Eisenbahnlinie von Fianarantsoa zur Ostküste. Die lokale Rattenpopulation ist oft mit dem Pesterreger in Kontakt gekommen (GOODMAN, mündliche Mitteilung). Forêt de Vinantelo Der Forêt de Vinantelo liegt mitten im Regenwaldgebiet des Ranomafana- Andringitra-Korridors auf ca. 1100 m Höhe. Die ersten zoologischen Lebendnachweise von Rattus rattus auf Madagaskar wurden Ende des 19. Jahrhunderts von Forsyth-Major erbracht. Die Fänge aus diesem Waldgebiet wiesen Kontakt mit dem Beulenpesterreger auf (GOODMAN mündliche Mitteilung). Südosten - Die Tolagnaro / Fort Dauphin Region: Die Region um Tolagnaro liegt im südlichsten Bereich des immergrünen Küstenregenwaldes der Ostküste Madagaskars. Der Küstenregenwald gilt als eigenes Ökosystem, welches sich in seiner Flora- und Faunazusammensetzung vom immergrünen Regenwald weiter im Inneren des Landes unterscheidet (LOWRY & FABER-LANGENDORN 1991; DUMETZ 1999; GANZHORN ET AL. 2000) und ursprünglich den Grossteil der Vegetation an der niederen Ostküste ausmachte (G REEN & S USSMAN 1990). Der Wald wächst hier auf sandigem Grund, erreicht eine Höhe von 18 m und weist einen dichten Unterwuchs auf (DU PUY & MOAT 1996; DUMETZ 1999). Um Tolagnaro sind etwa 4000 ha Küstenregenwald erhalten, wovon 1992 noch 87% als intakt galten (L EWIS - E NVIRONMENTAL -C ONSULTANTS 1992). Diese verbliebenen Fragmente des Küstenregenwaldes gehören zu den am stärksten bedrohten Habitaten Madagaskars (DU PUY & MOAT 1998; GANZHORN et al. 2001; RAMANAMANJATO & GANZHORN 2001). Der Niederschlag in der Region beträgt meist über 2000 mm pro Jahr, wobei die Gebiete Ste Luce und Mandena höheren Niederschlagsmengen ausgesetzt sind als Petriky, da der Südost-Passat mitgeführte Wassermassen vor dem Bergmassiv im Landesinneren abregnet (KOECHLIN 1972; DUMETZ 1999). Eine Trockenzeit gibt es nicht (RAMANAMANJATO & GANZHORN 2001). Die Temperatur liegt im Jahresmittel bei 26°C, sie nimmt von Norden nach Süden hin zu. In den Wintermonaten können Zyklone auftreten (DONQUE 1972; WWF 2001). Die klimatischen Unterschiede führen zu zwei Formen des Küstenwaldes: dem feuchten Küstenregenwald, wie er in Ste Luce und Mandena vorliegt und dem trockenen Küstenübergangswald, wie er in Petriky (Abb. 5) zu finden ist. Generell ist die Art der Vegetationszusammensetzung in dieser Region eher von der
2. Material und Methoden 11 Höhenlage als von der geographischen Breite des Habitats abängig (DONQUE 1972; DU PUY & MOAT 1996). Die Stadt Tolagnaro verfügt über einen eigenenen Überseehafen. Ste Luce Ste Luce liegt etwa 50 km nordöstlich von Tolagnaro. Etwa 43% des noch intakten Küstenregenwaldes sind hier zu finden (UNKNOWN & STEER 2000). Wie zuvor schon in Mandena soll von der Qit Madagascar Minerals (QMM) Titaneisen (FeTiO 3 ) aus den Sandböden gewonnen werden. Die gefangenen Tiere stammen daher sowohl aus intakten als auch aus degradierten Waldgebieten. Abb. 5: Die Tolagnaro Region (nach QMM 1998, DAWSON (2000) & INGRAM ET AL. (2000)) Mandena Das Untersuchungsgebiet Mandena liegt 12 km nordöstlich von Tolagnaro. Von dem ursprünglich zusammenhängenden Waldgebiet existieren lediglich noch Fragmente (Abb. 6), die jedoch alle kleiner als 1000 ha sind. Der Wald ist immergrün, 10-15 m hoch und hat einen dichten Unterwuchs. Die Fragmente M4 & M5 (Abb. 6) sind durch Sumpfgebiete mindestens seit 1957 von den restlichen Fragmenten isoliert. Zwischen M4 & M5 wurde 1998 ein künstlicher Korridor aus 80% exotischen und 20% einheimischen Bäumen angelegt. Daher wurden die Probenstellen beider Fragmente für die vorliegende Arbeit zusammengefasst.
2. Material und Methoden 12
Abb. 6: Primärwaldfragmente in Mandena, inclusive der Probenstellen M4, M5 und M16
(RAMANAMANJATO & GANZHORN 2001).
M16 war ursprünglich mit M20 verbunden, jedoch wurde der verbindende
Primärwald sukzessiv von Eucalyptus und Pinus Plantagen ersetzt. Rattus rattus
wurde sowohl in den Primärwaldgebieten als auch in sekundärer Vegetation
gefangen, während endemische Arten nur in Primärwaldgebieten zu finden waren
(zusammengefasst aus RAMANAMANJATO & GANZHORN (2001), siehe auch
LETHONEN et al. (2001)). Die in der Arbeit berücksichtigten Fragmente sind in
Tabelle 1 dargestellt.
Tab. 1: Fragmentgrössen und Degradationszustand in Mandena (RAMANAMANJATO & GANZHORN
2001).
Fragment Grösse [ha] Degradation
[in % des erhaltenen Blätterdaches]
M4/5 69 30-40
M16 75 602. Material und Methoden 13
Petriky
Die Beprobungsstelle Petriky liegt südöstlich von Tolagnaro. Da sie sich nicht
mehr im Einflussbereich der Regengebiete vor dem Bergmassiv befindet, sind die
Niederschläge deutlich geringer (DONQUE 1972). Der trockene Regenwald
repräsentiert die Übergangszone zwischen dem typischen Küstenregenwald
weiter nördlich und der xeromorphen Vegetation im Süden der Insel (CHAUVET
1972; GOODMAN et al. 1997). Die Kronenhöhe ist auf 10 m begrenzt (GOODMAN et
al. 1997; SCHAD 2001).
2.2.2 Probenmaterial
Aus den beschriebenen Regionen standen mir für die mtDNA
Untersuchung insgesamt 100 Proben von Rattus rattus zur Verfügung. Die
Proben stammten aus unterschiedlichen Projekten und wurden für diese
Untersuchung zusammengetragen. Die Tiere wurden mit ShermanTraps von den
in Tabelle 2 aufgeführten Biologen gefangen. Der Tabelle sind ebenfalls die
Anzahl der Proben pro Gebiet zu entnehmen. Gefangen wurde in den
verschiedenen Projekten insgesamt von 1998 bis 2001. Das Gewebematerial
bestand aus Ohrknorpel and wurde in Ethanol (70%) konserviert. Für die globale
Einordnung der madagassischen Ratten wurden zehn weitere Proben analysiert,
die mir aus Frankreich von J. Abdelkrim (Museum National d'Histoire Naturelle,
Paris), aus Gross-Britannien von S. Nakagawa (University of Sheffield) und aus
Französisch Polynesien von L. Matisoo-Smith (University of Auckland) zugesandt
wurden. Die Untersuchung wurde an der nichtkodierenden hypervariablen Region
(HVR1) des d-loop der mtDNA und am kodierenden Exon 2 des MHC DRB Gens
durchgeführt.
Tab. 2: Probenorte, Angabe der verwendeten Probenanzahl (N), Fangperiode, Proben
bereitgestellt von (Provider) und Vegetationstyp.
Probenort N Fangperiode Proben von Vegetationstyp
Analavelona 22 2000 Goodman (WWF Madagascar Regenwald
& Field Museum, Chicago)
Ioranjatsy 13 2000 Goodman (s.o.) Tavy -Wald
Vinantelo 12 2000 Goodman (s.o.) Montaner
Regenwald
Ste Luce 12 2001 Hapke (DPZ Göttingen) Küstenregenwald
Mandena M4/5 12 1998, 1999, 2001 Ganzhorn Küstenregenwald
(Universität Hamburg)
Mandena M16 13 1998, 1999, 2000 Ganzhorn (s.o.) Küstenregenwald
Petriky 8 1999, 2001 Ganzhorn, Rüdel, Schüller trockener Küsten-
(Universität Hamburg) übergangswald2. Material und Methoden 14 2.3 Molekulargenetische Methoden Die Analyse der mitochondriellen DNA (mtDNA) und die Analyse des Haupthistokompatibilitätskomplexes (MHC) wurde anhand unterschiedlicher molekulargenetischer Methoden durchgeführt. Nach der DNA-Extraktion wurde durch die Polymerasekettenreaktion (PCR) mit spezifischen Primern das jeweilige Fragment amplifiziert. Die Ergebnisse wurden durch Agarosegelelektrophorese kontrolliert. Die haploide mtDNA wurde anschliessend aufgereinigt und sequenziert. Die diploide MHC-DNA wurde über ein SSCP-Gel in ihre einzelnen Allele aufgetrennt, die Allelbanden ausgeschnitten, gereinigt, erneut amplifiziert und gereinigt und daraufhin ebenfalls sequenziert. 2.3.1 DNA Extraktion Die in Ethanol konservierten Gewebeproben wurden auf Papier getrocknet und anschliessend 1½ h in dH20 vom restlichen Ethanol gereinigt. Das erneut getrocknete Gewebe wurde nach dem Protokoll DNeasy™ for Animal Tissues von QIAGEN (04/99) weiter behandelt. Die Proben wurden in 1,5 ml Tubes überführt, die mit 180 µl ATL Buffer und 20 µl Proteinase K vorbereitet waren. Sie wurden mechanisch zerkleinert, anschliessend gevortext und bis zur vollständigen Lyse (mind. 3 h) unter ständigem Schütteln im Wasserbad bei 55°C inkubiert. Zur besseren Lyse wurde alle 30-60 min kurz gevortext. Nach kompletter Auflösung des Gewebes wurde 200 µl AL Buffer zu dem Lysat pipettiert und nach sorgfältiger Durchmischung 10 min bei 70°C inkubiert. Weitere Aktivität der Proteinase K wurde so unterbunden. Zur Fällung der DNA wurden daraufhin 200 µl Ethanol (96-100%) hinzugegeben, erneut sorgfältig durchmischt und die Lösung anschliessend in ein DNeasy Mini Column (in 2ml Collection Tube) überführt und bei 8000 rpm 1 min zentrifugiert. Der Durchfluss wurde verworfen. Die Silica-Säule mit der gefällten zu reinigenden DNA wurde nun in zwei Waschschritten (AW1 und AW2 Buffer) von restlichen Salzen und Ethanolresten gereinigt. Der Durchfluss wurde beide Male verworfen. Die MiniColumn mit der so getrockneten Membran wurde dann in ein 1,5 ml Tube umgesetzt und, abweichend der QIAGEN Anleitung, zur Eluation der DNA mit 200 µl dH2O versetzt, 5 min stehen gelassen und bei 8000 rpm 1 min zentrifugiert. Dieser Schritt wurde mit 150 µl dH2O wiederholt. Alle Eluate wurden zur quantitativen Überprüfung der DNA Extraktion über ein Agarose Gel elektrophoretisch aufgetrennt. Bei positivem Ergebnis wurden beide Chargen danach vereinigt. Zur Weiterverarbeitung wurden 50 µl als Arbeitsstock entnommen (Lagerung 4°C), die restlichen 300 µl als Reservestock bei -20°C aufbewahrt.
2. Material und Methoden 15 2.3.2 Agarose-Gelelektrophorese Die Produkte der DNA-Extraktion und der PCR (5 µl) wurden in 2%igen Agarose- Gelen elektrophoretisch aufgetrennt. Es handelt sich dabei um das Standardverfahren zur Trennung und Identifizierung von Nukleinsäuren. Agarose ist ein Polysaccharid, welches aus roten Meeresalgen gewonnen wird. Sie wurde in Elektrophoresepuffer (1xTAE) aufgenommen und dann durch Erhitzen in der Mikrowelle in Lösung gebracht. Nach Abkühlung auf ca. 60°C wurde das Ethidiumbromid hinzugegeben, gut vermischt und dieser Ansatz in einen mit Kämmen vorbereiteten Gel-Schlitten gegossen. Während des Erstarrens wird eine homogene Gelmatrix ausgebildet, die innerhalb des Gleichspannungsstromfeldes während der Elektrophorese die Auftrennung nach Molekülgrösse und Ladung ermöglicht. Die Grösse der Matrix ist abhängig von der verwendeten Agarose Menge. Das Gel wurde in die Gelelektrophoresekammer gelegt und vollständig mit TAE-Puffer bedeckt. Die DNA-Lösung wurde mit ca. 1 µl Loading Dye vermischt in die Taschen pipettiert. Ein DNA–Längenstandard (100bp Ladder) daneben aufgetragen ermöglichte die Abschätzung der DNA-Fragmentlänge. Die Laufzeit der Gele betrug bei 60-80V etwa 40-60min. Die Visualisierung der Nukleinsäuren erfolgte durch das Ethidiumbromid, welches mit den Basenpaaren der DNA interkaliert und somit das Fluoreszenzverhalten unter UV-Anregung verstärkt, so dass die angefärbten Molekülbanden sichtbar werden (KNIPPERS 1997). Das UV bestrahlte Gel wurde über einen Videoprinter dokumentiert. DNA Isolierung aus Agarose Gelen Einige Einzelbanden wurden zur Reamplifizierung oder Sequenzierung unter möglichst geringer UV-Beleuchtung, da sonst Pyrimidindimere induziert werden (WEHNER & GEHRING 1995), aus dem Agarosegel ausgeschnitten und anschliessend mit dem Gel Extraction Kit (Qiaquick) gereinigt. Hierfür wurden die ausgeschnittenen Banden eingewogen, das dreifache Volumen des QG-Buffers hinzugegeben und unter mehrfachem Vortexen 10 min bei 50°C inkubiert. Um einen pH-Wert von 7.5 einzuhalten, sollte gegebenenfalls NaAcetat hinzugegeben werden. Die Lösung wurde nach Zugabe des einfachen Volumens an Isopropanol in eine Spin Column überführt und 1 min bei 13000 rpm zentrifugiert. Für den folgenden Waschschritt der an der Säulenmembran fixierten DNA wurde 0,75 ml PE Buffer hinzugegeben und bei 13000 rpm 1 min zentrifugiert. Der Durchfluss der letzten beiden Schritte wurde verworfen. Anschliessend wurde die Säule in ein 1,5 ml Tube überführt und die DNA mit 30 µl dH2O 5 min von der Membran gelöst und 1 min herunterzentrifugiert (13000 rpm). Nach Wiederholung der Eluation mit gleicher Menge dH2O wurde das Gesamtvolumen auf der SpeedVac auf etwa die
2. Material und Methoden 16 Hälfte eingeengt und für grösstmögliche Reinheit eine Ethanolfällung durchgeführt. 2.3.3 PCR - Polymerase Chain Reaction Die Polymerase Kettenreaktion (PCR) ist eine Methode zur Vervielfältigung von spezifischen DNA-Fragmenten, die 1983 von K. B. Mullis erfunden wurde. Eine Abfolge von Denaturierung, Annealing und Elongation wird zyklisch wiederholt, wobei theoretisch in jedem Zyklus der zu amplifizierende Abschnitt verdoppelt wird, was zu einer exponentiellen (2n; n=Zyklenanzahl) Amplifikation führen müsste. Real wird jedoch nur ein Multiplikationsfaktor von ca. 1,6 bis 1,7 erreicht, da die Vermehrungsmenge am Anfang geringer ist und am Ende die Amplifikation durch Pyrophosphat, Nukleotidfragmente und rehybridisierendes Produkt gehemmt wird (MÜLHARDT 2001). Der PCR-Zyklus beginnt mit der Hitzedenaturierung (92°C) eines doppelsträngigen DNA-Fragmentes zu zwei einzelsträngigen Matrizen. Da die enzymatische Synthese ausschließlich in 5´Æ3´- Richtung verläuft, werden zwei regiospezifische, dem Anfangs- und Endpunkt des zu amplifizierenden Bereichs komplementäre Oligonukleotide (Primer) benutzt, die sich bei ca. 50-60°C an die beiden 3´-Enden dieses gewünschten DNA-Fragmentes anlagern (Annealing). An diesem kurzen doppelsträngigen Abschnitt kann eine thermostabile DNA Polymerase die Neusynthese des Stranges mit Hilfe der Desoxynukleotidtriphosphate (dNTP) beginnen (Elongation). Die Thermostabilität der Polymerase wird aufgrund der Automatisierung (programmierbare Thermocycler) des Gesamtablaufs benötigt. Das Temperaturoptimum für die meist benutzte Taq-Polymerase (nach Thermophilus aquaticus) liegt bei 72°C. Für eine optimale Aktivität des Enzyms werden ausserdem 1-3mM Mg2+-Ionen benötigt. Da die Aktivität der Taq ab 40 Zyklen ermüdet und auch unspezifische Amplifikate vermehrt auftreten, sollte die Zyklenanzahl darunter bleiben. Primer Für eine möglichst spezifische Amplifikation ist die Sequenz der Primer ausschlaggebend, sowie die sich daraus ergebende Schmelztemperatur. Diese hängt hauptsächlich von der Anzahl der Basenpaare und dem Anteil an G/C Paaren ab und kann in ihrer einfachsten Form wie folgt berechnet werden (MÜLHARDT 2001): Tm = 4 x (Anzahl G+C) + 2 x (Anzahl A+T) [°C]. Die normale Länge für Primer variiert zwichen 18 und 25 Basen. Ihr Anteil an Cytosin und Guanin sollte 40-60% betragen und es sollten nicht mehr als vier gleiche Basen nacheinander enthalten sein. Wichtig ist, dass die Primer intern
2. Material und Methoden 17 keine Sekundärstrukturen wie z. B. sogenannte ’hair pins’ bilden, und dass sie nicht miteinander hybridisieren. Für die Amplifikation der HVR I des d-loops der mtDNA wurden die Primer L283 & H16498 verwendet. Die Nummern zeigen die Position des 3’ Endes der Primer für die menschliche mtDNA Sequenz an (ANDERSON et al. 1981). L steht für ‘light strand’ und H steht für ‘heavy strand’. Bei Amplifikationsschwierigkeiten wurden die Vor- und Rückprimer mit je einem anderen Primer ergänzt. Hierfür wurden Eli-r und Eli-f eingesetzt, konstruiert von S. Sommer nach den oben beschriebenen Kriterien. Für die Amplifikation des MHC DRB Exon 2 wurden verschiedene Oligonukleotide eingesetzt. Das Primerpaar Tub1JS und Tub2JS (SCHAD et al. 2003) sollte auf Nullallele (einzelne Allele die mit einem Primerpaar aufgrund von Mutationen in der Primerbindungsstelle nicht amplifiziert werden) getestet werden. Hierfür wurde mit dem Degenerate-Primer MUFP1 (GO et al. 2002) amplifiziert. Es gilt: Y = C oder T und W = A oder T (nach IUB: International Union of Biochemistry and Molecular Biology). In Tab. 3 sind alle Primer mit ihren Sequenzen aufgelistet. Alle unmarkierten Primer wurden von der Firma MWG- Biotech AG in Ebersberg hergestellt, die mit * markierten bei der Firma Gibco BRL Life Technologies in Karlsruhe. Tab. 3: Primersequenzen für die Amplifikation und die Sequenzierung der HVR1 des d-loops und des DRB Exon2 Gens von R. rattus. Primername (Richtung) Sequenz (5´ to 3´) L283* (forward) TAC ACT GGT CTT GTA AAC C H16498* (reverse) CCT GAA GTA GGA ACC AGA TG Eli-f (forward) CGA ACC AAT CTT CTT AGG GCA TC Eli-r (reverse) CGT AGG AAG GAG ATG TCT GAT AAA G Tub1JS (forward) GAG TGT CAT TTC TAC AAC GGG ACG Tub2JS (reverse) GAT CCC GTA GTT GTG TCT GCA MUFP1 (forward) TGA GTG TCA YTT CYW CAA YGG GAC Die PCR-Bedingungen wurden an die verschiedenen verwendeten Primer durch Temperaturgradienten-PCR, Variationen im Magnesiumgehalt und den Annealing- zeiten angepasst. Alle PCR–Bedingungen, die in dieser Arbeit erfolgreich verwendet wurden, sind im folgenden aufgelistet. Verwendet wurden die Programme MTDNA für die Amplifikation der mtDNA, TOUCHDOWN für DRB Exon2 Amplifikation für den SSCP Einsatz und das Programm DRB für die Amplifikation vor der Sequenzierung der aus SSCP-Gelen isolierten Allele. Die entsprechenden Reaktionsansätze (MasterMix) sind in Tab. 4 dargestellt. Der Erfolg der PCR wurde jeweils durch eine Elekrophorese über ein 2%iges Agarose-Gel überprüft.
2. Material und Methoden 18 Programme auf Biometra-PCR-Geräten (TGRADIENT und TPERSONAL) Der Heizdeckel wurde jeweils auf 10°C höher als die Denaturierungstemperatur vorgeheizt. PCR für mtDNA (MTDNA) 1. Denaturierung 94,0°C 0m 45s 2. Annealing 50,0°C 0m 45s 3. Elongation 72,0°C 1m 0s │zu Schritt 1 ► 34Zyklen 4. Extension 72,0°C 5m 0s 5. Kühlung 4,0°C Pause PCR für DRB (vor SSCP) (TOUCHDOWN) 1. Denaturierung 94,0°C 2m 0s 2. " 92,0°C 0m 30s 3. Annealing 65,0°C 1m 0s │je Zyklus -1°C 4. Elongation 72,0°C 1m 0s │zu Schritt 2 ►10 Zyklen 5. Denaturierung 94,0°C 0m 30s 6. Annealing 55,0°C 1m 0s 7. Elongation 72,0°C 1m 0s │zu Schritt 5 ► 30 Zyklen 8. Extension 72,0°C 2m 0s 9. Kühlung 4,0°C Pause PCR für DRB (vor Sequenzierung) (DRB) 1. Denaturierung 92,0°C 1m 0s 2. Annealing 53,3°C 0m 30s 3. Elongation 72,0°C 1m 0s │zu Schritt 1 ► 34 Zyklen 4. Extension 72,0°C 2m 0s 5. Kühlung 4,0°C Pause
2. Material und Methoden 19 Tab. 4: Einzelne Reaktionsansätze mit spezifischer Einsatzmenge der unterschiedlichen Primer und des DNA-Templates. MasterMix für: MTDNA TOUCHDOWN DRB Ansatzmenge 20µl 10µl 20µl Incubation Buffer 2µl 1µl 2µl dNTP / ddNTP 1,6µl 0,8µl 1,4µl Primer1 L283 1,0µl MUFP1 0,5µl Tub1JS 0,3µl Primer2 H16498 1,0µl Tub2JS 0,2µl Tub2JS 0,3µl dH2O 13,5µl 5,4µl 13,8µl Taq-Poymerase 0,2µl 0,1µl 0,2µl DNA-Template 1µl 2µl 2µl 2.3.4 Ethanolfällung Vor allen Schritten, die eine möglichst reine DNA benötigten, wurde eine Ethanolfällung durchgeführt. Hierbei macht man sich die Eigenschaft der DNA zunutze, in Gegenwart von Salzen bei einem Überschuss an Alkohol aus der wässrigen Lösung auszufallen. Die etwa 20 µl PCR- oder Autosequence Produkte wurden hierfür mit 80 µl 0,3M NaAc (pH 5,2) und 300 µl eisgekühltem 96%igen Ethanols vermischt, 5 min inkubiert und dann 25 min bei 13000 rpm zentrifugiert. Für das anschliessende Dekantieren empfahl es sich, die Tubes in einer definierten Richtung zu zentrifugieren, um bei der Weiterverarbeitung das Lösen des kaum sichtbaren DNA-Niederschlages (Pellets) vom Boden zu vermeiden. Zum Auswaschen des restlichen Natrium-Acetats wurde nach Zugabe von 100 µl eisgekühltem 70%igen Ethanol kurz runterzentrifugiert und wieder dekantiert. Das restliche Ethanol wurde über die Vacuumzentrifuge abgezogen. Für die laborinterne Weiterverarbeitung wurde das Präzipitat in 20 µl dH2O aufgenommen. Die für die Sequenzierung bestimmte DNA wurde bis zur Abgabe im Service Labor des Instituts für Zellbiologie und Klinische Neurobiologie der Universität Hamburg trocken bei -20°C gelagert. 2.3.5 Single Strand Conformation Polymorphism (SSCP) SSCP ist eine einfache und sehr sensitive Methode, mit der die durch Nukleotidsubstitution einhergehende Konformationsänderung der einzelsträngigen DNA (ssDNA) dargestellt werden kann (ORITA et al. 1989). Bei geringer Temperatur und unter nicht denaturierenden Bedingungen falten sich die Stränge
2. Material und Methoden 20 der ssDNA sequenzspezifisch auf und zeigen im Polyacrylamid-Gel ein unterschiedliches Laufverhalten, das auf dieser gebildeten Tertiärstruktur beruht (LIU et al. 1999; SUNNUCKS et al. 2000). Anhand des daraus entstehenden charakteristischen Bandenmusters können die Genotypen mit ihren Allelen bestimmt werden. Die Empfindlichkeit der Methode verhält sich umgekehrt proportional zur Fragmentgrösse, wobei zwischen 100-300 bp ein Nachweis einer einzigen Basensubstitution noch mit 99%iger Wahrscheinlichkeit aufgelöst wird (ORTI et al. 1997). Anwendung Das dehydrierte Gel wurde für ca. 1h auf 40ml DELECT Puffer rehydriert, anschliessend mit Drying Cardboard sorgfältig getrocknet und auf die bereits auf Lauftemperatur (12°C) vorgekühlte Keramikplatte der horizontalen Elektrophoreseapparatur plan aufgebracht. Ein intermediärer dünner Kerosinfilm (ca. 1,5 ml) gewährleistet eine homogene Temperaturübertragung. Die Anode und Kathode flankierend, wurden die in Elektrodenpuffer (+/-) getränkten Elektrodenstreifen an das Gel gelegt und somit der Kontakt zwischen Gel und Elektroden hergestellt. Bei der gesamten Vorbereitung ist auf eine exakte Austarierung der Gerätschaften zu achten, sowie auf eine luftblasenfreie Verteilung der Flüssigkeiten. Je nach PCR-Ergebnis wurden 1-6 µl Produkt eingesetzt und mit 6-8 µl Loading Dye vermischt, anschliessend 10 min bei 50°C denaturiert und dann je 6 µl in die Geltaschen pipettiert. Die Elektrophorese startete unter Normalbedingungen für ein 36 Kammer-Gel mit einem Vorlauf von 20 min bei einer Spannung von 200V (20mA / 10W); der Hauptlauf erfolgte bei 450V (30mA / 20W). Es wurden verschiedene Gele mit verschiedenen Hauptlaufzeiten eingesetzt (Tab. 5). Tab. 5: Eingesetzte Gele mit deren Laufzeiten und dazugehörige Elektrodenstreifen mit Puffer Einsatzmenge "SSCP-Gel“ 10% CleanGel HP15% DELECT-Puffer-System DELECT-Puffer-System Glasfaser Elektrodenstreifen in 25-30ml Papier Elektrodenstreifen in 20ml Laufzeit des Gels: ca 3½h Laufzeit des Gels: ca 4-4½ h Anschliessend an die Elekrophorese wurde das Gel vom Kerosin gesäubert und zur Silberfärbung und Fixierung in den Hoefer Automated GelStainer überführt. Die Silberfärbung ist ein autokatalytischer Prozess in dem sich Silberionen (Ag+)
2. Material und Methoden 21
an die ssDNA anlagern und zu metallischem Silber (Ag0) reduziert werden, das
durch seine schwarze Farbe das Bandenmuster sichbar werden lässt. Die
Färbung wurde in sechs Schritten nach Herstellerprotokoll durchgeführt und ist in
Tab. 6 zusammenfassend dargestellt. In Step 5 wurde das Developing manuell
abgebrochen, um ein optimales Ergebnis zu erzielen. Das Gel wurde über Nacht
an der Luft getrocknet und konnte danach ausgewertet werden. Bei Bedarf wurden
einzelne Allele aus den kontaminationsfrei gelagerten Gelen ausgeschnitten und
konnten so einzeln sequenziert werden (SUZUKI et al. 1991).
Tab. 6: Protokoll der Silberfärbung
Step Prozess Reagenz Dauer
1 Fixing 40 ml Fixing Concentrate (5x) 40 min
160 ml Fixing Diluter (~24%EtOH)
2 Washing 100 ml Washing Concentrate (6x) 3x10 min
500 ml dH20
3 Silvering 40 ml Silvering Concentrate 40 min
160 ml dH20
260 µl Formaldehyd
4 Water 400 ml dH20 1x2 min
1x1 min
5 Developing 40 ml Developing Concentrate (5x) >7 min
160 ml dH20
260 µl Formaldehyd
200 µl Thiosulfate Concentrate
6 Stoppping & 40 ml Stopping Concentrate (5x) 1x30 min
Preserving 160 ml dH20
Isolierung aus SSCP Gelen und Reamplifizierung
Zur Sequenzierung einzelner Allele wurden die gefärbten Banden unter sterilen
Bedingungen aus dem SSCP Gel ausgeschnitten (SCHAD 2001). Unter Zusatz von
ca. 20 µl 1x TBE–Puffer wurden die zerkleinerten Gelstücke über Nacht
geschüttelt und so die Fixierung aufgehoben. Die folgende PCR wurde unter
Standardbedingungen (DRB vor Sequenzierung) durchgeführt.2. Material und Methoden 22
2.3.6 Sequenzierung
Die Sequenzierreaktion erfolgte enzymatisch nach der Kettenabbruch-Methode
(SANGER et al. 1977). Durch Zugabe eines geringen Anteils an Didesoxy-
nukleosidtriphosphaten (ddNTP) zu den dNTP´s wird dabei die Synthese des
neuen Stranges zufällig, aber basenspezifisch abgebrochen, so dass DNA
Fragmente aller möglichen Kettenlängen entstehen. Die Fragmente der
Sequenzierung werden anschliessend elektrophoretisch der Grösse nach
aufgetrennt. Durch die basenabhängige Fluoreszenzmarkierung der ddNTP´s ist
eine direkte Detektierung der Basensequenz möglich.
Folgende Sequenzierbedingungen wurden für die sogenannte AUTOSEQ Reaktion
verwendet:
Heizeckel auf 106°C
1. Denaturierung 96,0°C 0m 30s
2. Annealing 50,0°C 0m 15s
3. Elongation 60,0°C 4m 0s │zu Schritt 1) ► 26Zyklen
4. Kühlung 4,0°C Pause
Der Reaktionsansatz enthielt je Probe:
5 µl 2,5 x Puffer
9,0 µl dH2O
0,5 µl Primer (f oder r)
1,5 µl Big Dye
Es wurden standardmässig 4 µl PCR-Produkt eingesetzt, bei schwachen
Produkten wurde die Menge bis 10 µl Template variiert und die Gesamtmenge
durch den verminderten Einsatz von dH2O korrigiert. Das Sequenz Produkt wurde
mit Ethanol gefällt (siehe 2.3.4), anschliessend trocken und rückstandsfrei an das
Service Labor der Universitätsklinik Eppendorf gegeben. Die Auftrennung erfolgte
gelelektrophoretisch über den 377 DNA Sequencer von ABI Prism oder
kapillarelektrophoretisch am 3100 Genetic Analyser ebenfalls von ABI Prism. Die
chromatographischen Ergebnisse waren die Grundlage für die statistische
Auswertung.2. Material und Methoden 23
2.4 Auswertungsmethoden
Die chromatographischen Sequenzrohdaten wurden mit BioEdit (HALL 1999)
editiert und in GeneDoc (NICHOLAS et al. 1997) aligned. Die Varabilitätsindizes der
genetischen Diversität und der Nukleotiddiversität nach NEI (1987) wurden mit
dem Programm ARLEQUIN V 2000 (SCHNEIDER 2000) berechnet.
Für den populationsgenetischen Vergleich der verschiedenen Probenorte wurden
zwei Testverfahren angewandt. Zum einen wurde eine AMOVA (Analysis of
molecular Variance) durchgeführt: die AMOVA (EXCOFFIER et al. 1992) ist ein der
ANOVA (Analysis of Variance) analoger hierarchischer Ansatz, in dem die
Zuordnung von Unterschieden zwischen den Haplotypen auf verschiedenen
hierarchischen Ebenen als Analoga der FST (ΦST) genutzt wird (GONZÁLEZ et al.
1998). Die ΦST-Werte wurden (analog den FST-Werten) nach WRIGHT (1978)
interpretiert, so dass der Bereich von: 0 bis 0,05 eine geringe,
0,05 bis 0,15 eine moderate,
0,15 bis 0,25 eine große und
über 0,25 eine sehr große Differenzierung
charakterisiert. Als Unterscheidungs-Matrix für die Berechnung wurde TAMURA &
NEI (1993) gewählt, da neben der Unterscheidung zwischen Transitionen und
Transversionen bei den Transitionen auch Unterschiede zwischen Purin und
Pyrimidin Substitutionen berücksichtigt werden. Inzwischen hat sich
herausgestellt, dass es innerhalb der Hypervariablen Regionen nochmals
„Hotspots“ für Mutationsraten gibt, während die meisten Stellen eher eine
geringere Anzahl von Veränderungen zeigen (HASEGAWA et al. 1993; TAMURA &
NEI 1993; WAKELEY 1993; EXCOFFIER & YANG 1999). Es wird versucht diese
Heterogenität mit Hilfe einer Gamma Verteilung zu modellieren (UZZELL & CORBIN
1971), da eine Vernachlässigung der Unterschiede zu einer falschen
Einschätzung der genetischen Distanzen zwischen Sequenzen (HASEGAWA et al.
1993; TAMURA & NEI 1993) und somit zu einer falschen Auflösung der
genealogischen Strukturen führen kann (FINNILÄ et al. 2001; ENDICOTT et al. 2003).
Daher wurde für die Varianzanalyse die von EXCOFFIER & YANG (1999) für die
menschliche HVR I des d-loops ermitteltete Gamma Korrektur von a = 0,39
angewendet. Neben der AMOVA wurde der ’Exakt-Test für
Populationsdifferenzierung basierend auf den Haplotypenfrequenzen’ nach
RAYMOND & ROUSSET (1995) angewandt. Der Exakt–Test ist ein
Unabhängigkeitstest, der die beobachtete Verteilung mit einer Zufälligen
vergleicht. Die AMOVA und der Exakt-Test wurden mit ARLEQUIN V 2000
(SCHNEIDER 2000) berechnet.Sie können auch lesen

















































