Patienteninformation Dilatative Kardiomyopathie (DCM, CMD) Orphanet (www.orpha.net): ORPHA217604 - UKM
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
ERN Guard Heart Patienteninformation Dilatative Kardiomyopathie (DCM)
Patienteninformation
Dilatative Kardiomyopathie
(DCM, CMD)
Orphanet (www.orpha.net): ORPHA217604
1. Das normale, gesunde Herz Mit der Erweiterung der Herzkammern ist eine
Störung der Pumpfunktion verbunden, die zu
Das Herz ist ein muskuläres Hohlorgan, das durch Blutrückstau und klinischen Beschwerden der
eine Herzscheidewand in eine rechte und eine linke Herzinsuffizienz führt. Dies kann zur Flüssigkeits-
Hälfte unterteilt ist. Die rechte Hälfte übernimmt ansammlung in den Lungen, in den Knöcheln, im
den Bluttransport zwischen Herz und Lunge Unterbauch und anderen Organen, sowie zum
(Lungenkreislauf), die linke zwischen Herz und Gefühl der Atemlosigkeit bei Anstrengung oder gar
übrigem Körper (Körperkreislauf). in Ruhe führen. Diese Reihe von Beschwerden ist als
Herzversagen bekannt. In den meisten Fällen
Beide Herzhälften sind durch Herzklappen unterteilt
entwickelt sich DCM langsam, deshalb kann ein Herz
in einen Vorhof (Atrium) und eine (Haupt-)Kammer
im Herzultraschall bereits relativ stark betroffen
(Ventrikel). Um das Blut zu pumpen (Vorhof >
sein, bevor die DCM diagnostiziert wird.
Hauptkammer > Kreislauf), schlägt ein gesundes
Herz in Ruhe 50-100 mal pro Minute. Ausgelöst und
Bei den Patienten mit DCM besteht je nach Krank-
kontrolliert wird der Herzschlag durch einen
heitsausprägung eine Neigung zu schnellen Herz-
elektrischen Impuls im Sinusknoten (Herzschritt-
rhythmusstörungen der Vor- und Hauptkammern,
macher), der anschließend zu den Vorhöfen und
die schlimmsten Fall zum Schlaganfällen, Embolien
dann über AV-Knoten, His-Bündel und Purkinje-
oder Kreislaufstillstand führen können.
Fasern auf die Kammermuskulatur geleitet wird, die
sich dann organisiert zusammenzieht und so das Das Ruhe-EKG, aber auch der Herzultraschall zeigen
Blut in die beiden Kreisläufe pumpt. Nach jeder oft Auffälligkeiten.
Herzerregung und rhythmischen Muskelkontrak-
tion bildet sich die elektrische Erregung zurück und
die Kammermuskulatur entspannt sich, um sich
erneut mit Blut zu füllen.
Mittels EKG und Herzultraschall können diese
Vorgänge und die Anatomie des Herzens in Ruhe
oder unter körperlicher Belastung beurteilt werden.
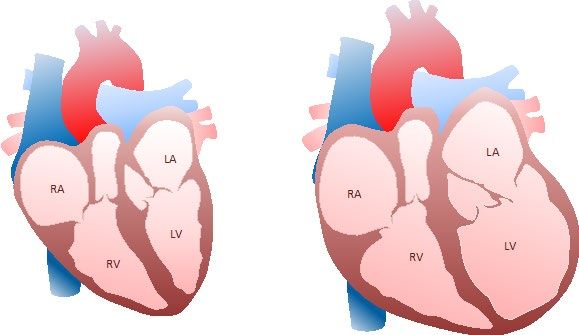
2. Dilatative Kardiomyopathie (DCM)
Unter der Diagnose „Dilatative Kardiomyopathie” Links: das normale Herz. Rechts: Herz mit DCM. R, rechts; L, links;
A, Atrium (Vorhof, Vorkammer), V, Ventrikel (Hauptkammer).
(DCM/CMD) wird eine Erkrankung des Herzens (=
Kardiomyopathie) mit Vergrößerung bzw. Erweite- 3. Häufigkeit und Vererbung der DCM
rung der Herzkammern (= Dilatation) verstanden.
Die DCM ist relativ selten, d.h. ca. jede 500 - 1.000
Zunächst ist in der Regel die linke Hauptkammer in nicht-verwandte Person ist betroffen (sog. Präva-
Form einer Erweiterung und später mit einer Pump- lenz oder Fallanzahl). Es handelt sich bei der DCM zu
schwäche (= Herzinsuffizienz) betroffen; bei fort- 40-50% um eine Erbkrankheit, d.h. Ursache ist
schreitender Erkrankung kann auch die rechte Herz- genetisch bedingt und an Nachkommen vererbbar.
kammer ähnlich betroffen sein. Meist sind mehrere Familienmitglieder, jedoch
Januar 2021 1 /5
ERN Guard Heart Patienteninformation Dilatative Kardiomyopathie (DCM)
individuell unterschiedlich in der Krankheits- Die meisten genetischen DCM-Unterformen (> 20
ausprägung und Symptomen, betroffen. verschiedene) sind autosomal (= geschlechts-unab-
hängig) dominant vererbt, d.h. das Vorkommen ei-
Die DCM ist im Kindes- und Jugendalter die häufig-
nes veränderten DCM-Gens* (bzw. einer Mutation)
ste Form der Herzmuskelerkrankung. Sie tritt u.a.
reicht aus, um die Erkrankung zu verursachen.
familiär gehäuft und ist dann genetisch bedingt,
kann aber auch durch eine Reihe äußerer Faktoren Seltener sind autosomal-rezessive oder digene
(z.B. Herzmuskelentzündung/Myokarditis, Durch- Formen (zwei Mutationen vorliegend; < 5%) oder
blutungsstörungen) sowie bei neuromuskulären Neumutationen (beide Eltern ohne Mutation; < 1%).
und Stoffwechsel-Erkrankungen auf. Die Erkran-
Nachkommen ohne die familiäre Genmutation
kung ist dann „erworben“ (sekundär, exogen).
(‘gesund’) können die Erkrankung nicht an ihre
Trotz erheblicher Fortschritte auf dem Gebiet der Nachkommen weitergeben bzw. vererben.
genetischen Diagnostik in den letzten Jahren ist
Typischerweise sind die DCM-assoziierten Symp-
diese bei Kindern und Jugendlichen unter 18 Jahren
tome, d.h. die Krankheitsausprägung der jeweiligen
nur bedingt (bei isolierter DCM nicht) notwendig.
Mutationsträger, innerhalb einer Familie unter-
Wenn die DCM im Rahmen einer neuromuskulären
schiedlich (sog. variable Krankheitsexpressivität)
Erkrankung auftritt oder z.B. zusätzlich eine Störung
und von zusätzlichen Faktoren abhängig.
des Reizleitungssystems besteht, ist eine genetische
Diagnostik sinnvoll und zumeist aufklärend. Bei ca. 50% der DCM-Patienten kann keine gene-
tische oder anderweitige Ursache festgestellt wer-
Mutationen, d.h. krankheitsverursachende Verän-
den. Man spricht dann von einer „idiopathischen“
derungen in der Erbinformation (DNA-Sequenz) von
DCM.
DCM-ursächlichen Genen, finden sich immer bei
allen Betroffenen einer Familie und sind für diese 4. Symptome bei DCM
jeweils spezifisch (“Jede Familie hat ihre eigene
Patienten, die an einer DCM erkrankt sind, können
Mutation”). Jedes DCM-Gen ist immer 2x im Erbgut
u.U. körperlich gut belastbar sein und relativ be-
vorhanden; die Wahrscheinlichkeit, dass ein verän-
schwerdefrei sein, wenn die Erkrankung z.B. früh im
dertes DCM-Gen* von dem erkrankten Elternteil an
Herzultraschall erkannt (und ggfs. behandelt)
einen Nachkommen weitergegeben wird, ist damit
wurde. Manche Patienten haben kaum Symptome,
rein statistisch (“gemittelt”) 50%.
wohingegen andere deutliche Probleme (s.u.) ent-
wickeln, welche eine komplexe Behandlung
erfordern.
Wenn Patienten mit DCM an Symptomen leiden,
können diese oft durch Medikamente oder andere,
zusätzliche Therapien behandelt werden. Die Symp-
tome von DCM sind ähnlich einem Herzversagen,
d.h. der Herzmuskel kann nicht mehr genügend Blut
durch Lungen und Körper pumpen. Hierzu gehören
u.a. Kurz- oder Schweratmigkeit unter Belastung
oder gar in Ruhe, Anschwellen der Knöchel, Füße
oder gar vom Unterbauch/Rücken, Müdigkeit und
fehlende Belastbarkeit, sowie Herzklopfen, Herz-
rhythmusstörungen; letztere können Vorhof-
Stammbaum oben: Vater (eckig, schwarz) erkrankt; Tochter #1
flimmern sein, andere Herzrhythmusstörungen sind
(rund, schwarz), Sohn #1 (eckig, schwarz) erkrankt und
Sohn #2 (eckig, grau) leicht erkrankt. Gesund sind die mitunter lebensbedrohlich, insbesondere wenn die
Mutter (rund, weiß) und Tochter #2 (rund, weiß). Pumpleistung des Herzmuskels deutlich einge-
Stammbaum unten: Mutter (rund, schwarz) erkrankt; Tochter #1 schränkt ist.
(rund, grau) leicht erkrankt, Sohn #1 (eckig, weiß) und
Vater gesund, Tochter #2 (rund, schwarz) und Sohn #2
(eckig, schwarz) erkrankt.
Januar 2021 2 /5ERN Guard Heart Patienteninformation Dilatative Kardiomyopathie (DCM)
5. Diagnose der DCM Blutdruck und Herzrhythmusstörungen sowie QT-
Intervall werden gemessen. Bei Patienten mit DCM
Die Diagnose einer DCM ist zunächst auf dem gibt das Belastungs-EKG Informationen über die
Herzultraschall beruhend, wo Herzfunktion und derzeitige Leistungsfähigkeit (+/- Therapie), Durch-
Herzkammergrößen beurteilt werden. blutungsstörungen und mögliche, belastungs-ab-
Bei auffälligen Befunden wird mitunter eine hängige Herzrhythmusstörungen.
zusätzliche Bildgebung (Kardio-MRT, mit Kontrast-
5.3 Das Langzeit-EKG (Holter-EKG)
mittelgabe) oder eine invasive Herzkatheter-
untersuchung (Darstellung der Herzkrankgefäße, Das Langzeit-EKG (Aufzeichnungsdauer: 1-7 Tage)
Kreislaufdruckmessungen) erforderlich. Mit einer ist ein kleiner EKG-Apparat (mit Umhängegürtel),
Blutentnahme können weitere Ursachen einer DCM wo die elektrische Herzaktivität mit 4 oder 6
(z.B. Stoffwechselerkrankungen, Herzmuskelent- Elektroden (am Brustkorb geklebt) aufgezeichnet
zündung, etc.) erkannt werden. werden. Es geht darum, schnelle oder langsame
Herz-rhythmusstörungen zu dokumentieren und
Die eigene Krankengeschichte (Anamnese), Fami-
nach Möglichkeit mit Symptomen zeitlich zu
lienanamnese (plötzlicher Herz-/Kinds-/Badetod,
korrelieren (Patienten-/Aktivitätsprotokoll).
andere Familienmitglieder mit DCM oder Defibril-
lator) sowie die körperliche Untersuchung wie die 5.4 Der Event-Recorder (Ereignisschreiber)
gezielte kardiologische Untersuchung können hin-
Es handelt sich um ein besonderes Langzeit-EKG mit
weisend für ein DCM sein.
der Aufzeichnungsdauer von bis zu 30 Tagen
*** Manchmal, insbesondere im frühen Stadium, (externer Event-Recorder/EKG-Apparat) oder um
kann die Diagnose schwierig sein. Manchmal ist die ein kleines Gerät, welches in das Unterhaut-
Herzkammer noch nicht wesentlich vergrößert, fettgewebe (Brustkorb links) für 1-2 Jahre einge-
jedoch etwas in der Pumpfunktion beeinträchtigt setzt wird (implantierter Loop-Recorder,
(„hypokinetisch“) oder auffallend von Herzrhyth- telemetrisch). Bei Symptomen kann über einen
musstörungen oder Extraschlägen betroffen. Diese Knopf das Gerät aktiviert werden, um mögliche
Phase wird auch als „Arrhythmogene (unklas- Herzrhythmusstörungen zu dokumentieren und
sifizierte) Kardiomyopathie (A(U)CM) bezeichnet. i.W. spezifisch zu behandeln. Es handelt sich um
eine Spezialdia-gnostik im Einzelfall.
5.1 Das Ruhe-EKG
5.5 Der Herzultraschall (Echokardiographie)
Über Elektrodenpflaster (6x Brustkorb, 2x Arme, 2x
Beine), die über Kabel an das EKG-Gerät Im Herzultraschall (Ruhe oder auch Stress-
angeschlossen sind, wird die Herzaktivität der untersuchung) werden Pumpfunktion und wichtige,
Vorhöfe und Hauptkammern in 12 verschiedenen anatomische Strukturen des Herzens (Kammern,
Ableitungen hochauflösend und kontinuierlich Herzklappen, herznahe Gefäße, Herzbeutel) beur-
registriert und neben der Herzfrequenz, dem teilt. Die Herzkrankgefäße sind nicht erkennbar.
Herzrhythmus (Extraschläge ja/nein, Herzrhythmus-
*** Weitere kardiologische Untersuchungen (Kar-
störungen ja/nein) auch Herzzeitintervalle (z.B. QT-
dio-MRT, Herzkatheter, Elektrophysiologische
Intervall) und das Aussehen einzelner EKG-
Untersuchung, Herzmuskel-Biopsie) sind meist
Komponenten (z.B. T-Welle, elektrischer Lagetyp
erforderlich.
des Herzens) beurteilt.
5.6 Genetische Untersuchungen
Bei Patienten mit DCM können im Ruhe-EKG
Vorhofflimmern, AV-Blöcke, Extraschläge aus den Genetische Untersuchungen werden am Erb-
Hauptkammern oder gar Herzrhythmusstörungen material (DNA) durchgeführt, was aus Blut (oder
aufgezeichnet werden. Körpergewebe) isoliert wurde. Ziel ist es, die Erbin-
formation des Patienten in Genen für die Erkran-
5.2 Das Belastungs-EKG kung “zu lesen”, um mögliche Abweichungen von
der Natursequenz zu detektieren; diese können
Es handelt sich um ein 12-Kanal-EKG unter Be-
entweder krankheitsverursachend (=Mutation)
lastung (meist Ergometer/Fahrrad); Herzfrequenz,
Januar 2021 3 /5ERN Guard Heart Patienteninformation Dilatative Kardiomyopathie (DCM)
oder auch nur einfache “natürliche Varianz” (Poly- Ein moderates Ausdauertraining kann im Hinblick
morphismus, ohne Krankheitsbedeutung) sein. auf Belastbarkeitssteigerung und Herzmuskelstär-
kung positiv wirken; bei reduzierter Pumpfunktion
Für jede Familie gibt es dabei eine individuelle, d.h.
ist eine kompetitive, hochintensive Aktivität unter-
für die Familie spezifische Genveränderung. Selten
sagt, freizeitsportliches Ausdauertraining ist jedoch
liegen 2 Genmutationen vor.
möglich. Bei DCM-Genträgern ohne klinische Mani-
Für das DCM sind >15 verschiedene Gene bzw. festation sind (fast) alle Sportarten möglich.
genetische Unterformen bekannt. Dennoch ist die
Aufklärungsrate mit einem positiven Mutations- 8. Regelmäßige Arztbesuche
befund (“Sensitivität”) nicht 100%, sondern ca. 30-
40%. Ein negativer Genbefund (ohne Mutations- Eine spezialisierte Einrichtung bzw. Zentrum wird
nachweis) schließt daher die Erkrankung nicht aus. Sie gemeinsam mit Ihrem Herzspezialisten und
Das DCM-Hauptgen ist das Gen TTN (Titin). anderen Ärzten vor Ort betreuen und Sie je nach
Bedarf (z.B. Symptome, Alter, spezielle Umstän-
Die genetischen Untersuchungen dauern ca. 2-8 de/Situationen/OP, Behandlungen) beraten, wie
Wochen, je nach Untersuchungsumfang/-art und häufig Vorstellungen und ggf. eine Anpassung der
sollten von einer humangenetischen und kardiolo- Therapie erforderlich sind. Auch werden Fragen
gischen Beratung begleitet werden. zum Lebensstil, der Einfluß auf die Erkrankung
haben kann, detailliert besprochen.
6. Therapie
Bei Bedarf kann ein Kontakt zu Selbsthilfegruppen
Eine ursächliche Therapie für eine DCM gibt es
oder auch zu anderen Ärzten hergestellt werden,
nicht; dennoch sind die frühzeitige Erkennung der
um eine umfassende und interdisziplinäre
Erkrankung und medikamentöse Therapie sinnvoll,
Betreuung sicher zu stellen.
um Symptome und den langfristigen Verlauf günstig
zu beeinflussen. Falls Patienten von einem DCM-Mutationsträger ohne aktuelle Zeichen der
plötzlichen Herztod bedroht sind (z.B. bei Erkrankung sollten sich in größeren Abständen (z.B.
schwerwiegender Herzinsuffizienz oder alle 3-5 Jahre) oder beim Auftreten erstmaliger
Herzrhythmusstörungen, die sich mitun-ter Symptome klinisch untersuchen lassen.
gegenseitig bedingen), kann die Behand-lung mit
einem Defibrillator (ICD, S-ICD) mit oder ohne 9. DCM und Familie
Herzschrittmacherfunktion notwen-dig sein. Bei
schwerer Herzinsuffizienz und Pumpschwäche Wurde eine DCM diagnostiziert und ist eine
können neben der intensiv-medizinischen angeborene Form wahrscheinlich, sollten unmittel-
Behandlung auch eine Behandlung mit einem bare (biologisch verwandte) Familienmitglieder –
Kunstherz bzw. eine Herztransplantation unabhängig von Symptomen - auf das Vorliegen
erforderlich sein. Bei ausgeprägter DCM besteht eines DCM untersucht werden (Ruhe- und
u.a. das Risiko der Entstehung von Thromben und Belastungs-EKG). Ist eine DCM-Genmutation be-
Embolien, das durch sog. Gerinnungshemmer/Blut- kannt, sollten sich die Familienmitglieder im Umfeld
verdünner gesenkt werden kann. einer humangenetischen und kardiologischen
Beratung diesbezüglich untersuchen lassen (sog.
7. Lebensstil und Sport Heterozygotentestung). Die Untersuchung ist dabei
entweder diagnostisch oder prädiktiv. Wird die
Durch Lebensstilmodifikation können bei Patienten bekannte DCM-Genmutation der Familie nachge-
mit DCM der klinische Verlauf, insbesondere die wiesen (‘positiver Gentest’)(Stufe 1: Eltern, Kinder
Herzinsuffizienz, in frühen und mittleren Stadien und Geschwister des Ersterkrankten der Familie),
der Erkrankung positiv beeinflusst werden. Die sollten im Rahmen der sog. Kaskadenuntersuchung
Stärke der Empfehlungen, den Lebensstil weitere Familienmitglieder (z.B. Kinder eines
anzupassen, als auch die der Therapie orientiert sich Bruders mit positivem Gentest) ebenfalls
am Ausmaß der Erkrankung. kardiologisch und genetisch untersucht werden
(Stufe 2, 3, …).
Januar 2021 4 /5ERN Guard Heart Patienteninformation Dilatative Kardiomyopathie (DCM)
Da DCM-Patienten bereits in der Kindheit oder nach
der Geburt Symptome haben können, ist eine
frühzeitige Diagnostik wichtig.
DCM-Familienangehörige mit der DCM-Genmuta-
tion (‘positiver Gentest’) werden auch als Muta-
tionsträger bezeichnet.
Die Untersuchung ist dabei entweder diagnostisch
oder prädiktiv (keine Erkrankungszeichen oder
Symptome) und unterliegt dem sog. Gendiagnostik-
Gesetz (GenDG).
Familienangehörige mit negativem Gentest (keine
DCM-Genmutation) sind bei gleichzeitigen,
unauffälligen EKG-Befunden diesbezüglich nicht-
betroffen; ihre Nachkommen wie auch Kontakt:
Angeheiratete müssen nicht weiter in Bezug auf
eine DCM untersucht werden, da keine Weitergabe- Spezialambulanz für Patienten mit genetischen
wahrscheinlichkeit besteht. Herzerkrankungen
Institut für Genetik von Herzerkrankungen (IfGH)
10. DCM und Schwangerschaft
(Leitung: Univ.-Prof. Dr. med. E. Schulze-Bahr)
Es ist sinnvoll, im Vorfeld einer Schwangerschaft
Universitätsklinikum Münster (UKM)
Ihre Fragen, potentielle Risiken, ggf. eine Umstel-
lung der Medikation (? embryotoxisch) und die E-Mail: herzgenetik@ukmuenster.de
interdisziplinäre Beratung und Art der Betreuung T. +49-251-83 44945
während der Schwangerschaft zu besprechen. F. +49-251-83 52980
Auch während der Schwangerschaft und im ersten https://www.ukm.de/index.php?id=swgh_uebersicht
Jahr hiernach sollten Verlaufsuntersuchungen statt-
finden.
11. DCM und offene Fragen
Bitte rühren Sie sich diesbezüglich.
Notizen:
https://guardheart.ern-net.eu
Januar 2021 5 /5Sie können auch lesen

















































