Alpha 1 Antitrypsinmangel - N.Kaufmann - medONLINE.at
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

Alpha‐1‐Antitrypsinmangel
N.Kaufmann
Welche Bedeutung hat Alpha‐1‐Antitrypsin? • Das Schutzenzym Alpha1‐Antitrypsin steht aufgrund eines Gendefektes und reduzierter Expression aus den Leberzellen für den Schutz der fragilen Lungenalveolen nicht zur Verfügung. Makrophagen und neutrophile Granulocyten bauen exogene Schadstoffe ab. Die freigesetzte „neutrophile Elastase“ wird durch die Antiprotease „alpha‐1‐Antitrypsin“ neutralisiert. Bei Dysbalance dieses Proteasen/Antiproteasen Gleichgewichtes werden die elastischen Fasern der Lunge zerstört, mit der Folge eines primären (congenitalen) Lungenemphysems. 09.01.2023
Alpha‐1‐ Antitrypsinmangel
eine „rare disease“
• Eine Domäne der Pneumologen
• AATD als „underdiagnosed disease“
• Andere Fachdisziplinen behandeln bis zur
Diagnosestellung Pat. mit AATD
(meist Ärzte für Allgemeinmedizin, Internisten, Nephrologen,
Dermatologen und Kardiologen)
09.01.2023
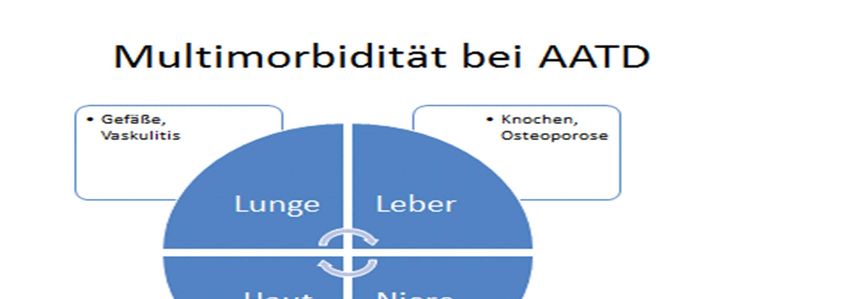
Wie wirkt sich der Mangel aus? • Dramatische Verschlechterung der Lungenfunktion schon in jüngeren Jahren (bei Nikotinabusus oder inhalativen exogenen Schadstoffen) durch Lungenemphysem, Bronchiektasen und chronische Inflammation • Hepatopathie (Alpha1‐Antitrypsinmangel ist de facto primär eine Lebererkrankung durch Akkumulation von fehlgefaltetem Alpha‐1‐ Antitrypsin in den Hepatozyten – Zielorgan ist jedoch die Lunge) 09.01.2023
Inflammation • Das Z Alpha‐1‐AT, welches in die Lunge diffundiert oder durch das Bronchialepithel und Monocyten produziert wird, kann Polymere innerhalb der Alveolen und der kleinsten Atemwege formieren und diese werden von Neutrophilen abgeräumt. • Diese Kaskade der Inflammation ist möglicherweise charakteristisch für Pat. mit AATD (David Lomas Cambridge) • LTB4, IL6 und IL8 erhöht bei bakteriellem Infekt 09.01.2023
Historisches, Forschung, Evidenz • Von der Detektion des „ Wikinger‐Gens“ bis zur Substitution 09.01.2023
AAT ‐ Chronologie • vor 2000 Jahren: Z‐Punktmutation in der skandinavischen Bevölk. • 8‐10 JH: Verbreitung des Z Gens durch die Wikinger in Europa • 1838: Ätiologie des Emphysems nach Laennec (chron. Bronchitiden) • 1955: path. Papierelphor. mit Kasuistiken nach Jacobsen • 1958: Emphysem entsteht durch destruktive Prozesse (Wright) • 1960: Antiobstruktive Therapie in Analogie zur COPD • 1961: primäres Lungenemphysem bei jungen Erw. (Schneider) • 1962: AFO durch Reduktion des „ elastic recoils“ (Laws) • 1962: Rolle des Kollagens und Elastins beim LE (Pierce) 09.01.2023

AATD ‐ Chronologie
• 1962: Isolierung des Glykoproteins AAT (Schultze, Behring Marburg)
• 1963: AATD medizinische Bedeutung (Carl Bertil Laurell)
• 1964: Erster Bericht der hereditären Erkrankung AATD
• 1969: Lebererkrankung bei AATD (Sharp)
• 1970: Assoziation zwischen AAT und neutrophiler Elastase (Travis)
• 1972: Assoziation zwischen AATD und Panniculitis (Warter)
• 1980: Erste Substitutionsversuche ( Gadek, Crystal NJH CC)
• 1992: Klinische Anwendung (Prolastin‐C, Aralast, Zemaira, Glassia)
• 1980: WATL (ARGE Th. Lungenerkr.) Arbeitsgruppe AATD, Deutsches Register
• 1984: Kristallisation AATD‐aktives Zentrum‐target enzym (R.Huber)
• 1985: LUTX
• 1997‐2005: Klinische Studien der Substitutionstherapie (Soersholm, Dirksen,
Wencker, Chapmann, Stockley, Liebermann)
09.01.2023
AATD ‐ Chronologie
• 1996: AIR (Alpha‐1 International Registry) 5000 p, 17c
• 1997: U.K. registry unter ADAPT ( Antitrypsin Deficiency Assessment
and Programm for Treatment)
• 1999: Polymere und inflamm. Folgen (Lomas)
• 2007: Bronchiektasen zu 27 % in PiZZ Pat. UK cohort , Parr
• 2009: „lung densitometry“ RCT, Dirksen
• 2011: Österreichisches Alpha‐1‐Register (Schmid‐Scherzer)
• 2014: Schnelltest AlphaKit Quick Screen ( Chiesi)
• 2014: RAPID‐Studie (pub. in Lancet)
06/2019 N. Kaufmann
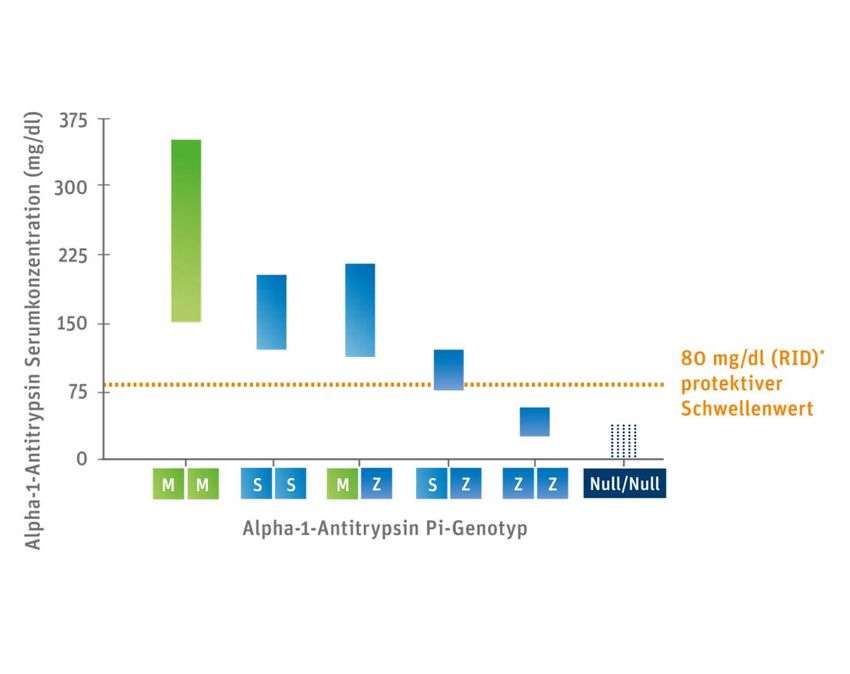
Welche genetische Veränderung liegt vor?
• Mehr als 100 verschiedene Allele des SERPINA Gens, das für Alpha‐1‐
Antitrypsin codiert, sind bekannt.
• Die häufigsten und klinisch relevanten sind
ZZ, MZ und SZ Varianten
• Die Normvariante ist das M‐Allel
• Auch eine 0‐Variante liegt vor (< 1%)
06/2019 N. KaufmannWie ist die Vererbung? Wo ist der Defekt? • Autosomal kodominant • Spontanmutation im Bauplan des Alpha‐1‐Gens (Chromosom 14) • Das Glykopeptid AAT besteht aus 394 Aminosäuren mit komplexer globulärer Tertiärstruktur (aktives Zentrum Methionin) • Glutamin wird durch Lysin an Position 342 ersetzt (Z‐Allel) • Wird in den Hepatozyten produziert, sezerniert und in die Blutbahn abgegeben. • HWZ 5 Tage • Syntheserate 34 mg/kg/die • Serumspiegel 90‐220 mg/dl 09.01.2023
Prävalenz –AATM/100.000 Einwohner (Orphanet Report Series, Prevalence of rare disease Bibliographic data 2014) 09.01.2023
Prävalenz in Österreich • Die österreichische Bevölkerung trägt zu 0,1% ein defektes Allel und weist meist ein heterozygotes Muster auf. • Nur 10 % dieser 0,1 % mit AAT‐Gendefekt sind klinisch manifeste homozygote AATM‐Patienten. • In Österreich ca. 2500 Patienten • Bisher 385 im Österreichischem Alpha‐Register erfasst • Hohe Dunkelziffer 09.01.2023
Welcher Spiegel ist ausreichend? 09.01.2023
Alpha‐1 Antitrypsinmangel • Ist eine genetische Erkrankung mit Systemcharakter durch eine ständige neutrophile Inflammation mit Zytokinproduktion mit rascherer Verschlechterung der Lungenfunktion bei einer bakteriellen Infektion im Vergleich zur COPD (Robert Stockley, Birmingham) 09.01.2023
Primäres Lungenemphysem
• Haupterscheinungsbild mit
panlobulärem basal betonten
Lungenemphysem, respiratorischer
Partialinsuffizienz durch
Diffusionsstörung in der BGA,
erhöhtes Residualvolumen und
reduzierte Einsekundenkapazität in
der Spirometrie sowie Reduktion
der Lungendichte im CT.
06/2019 N. KaufmannAlpha‐1‐Antitrypsinmangel
06/2019 N. KaufmannPrognostische Faktoren bei COPD und AATM
• Lungenfunktion
• Alter
• BMI
• Leistungsfähigkeit
• Inflammation
• Komorbiditäten
• Psychosoziale Situation
06/2019 N. KaufmannBronchiektasen als Inflammationstrigger
• Klinisch signifikant bei Befall von >4 Lungensegmenten mit
Symptomatik
• 28 % Bronchiektasennachweis nach Kohortenstudie UK (Par)
• 14% zentrale und 24 % periphere Bronchiektasen in der NHLBI
Obduktionsstudie.
• Keimspektrum im Vergleich zur COPD bei AATD ungünstiger. Im
Sputum und BAL IL8, TNF alpha und neutrophile Elastasen vermehrt.
06/2019 N. KaufmannWeitere Erscheinungsbilder
06/2019 N. KaufmannSymptomatik • Leitsymptom Dyspnoe • Reduzierte Lungenfunktion und COPD Symptomatik schon in jungen Jahren • Lebermanifestation • Hauterscheinungen 09.01.2023
Wann daran denken? Indikation zur quantitativen AAT‐Bestimmung (ATS/ERS‐Empfehlungen) • Fehlen der Alpha‐1‐Bande in der Serumelektrophorese • Früh aufgetretenes Lungenemphysem • Bronchopulmonale Symptomatik bei mehreren Familienmitgliedern mehrerer Generationen • Lebererkrankung unklarer Genese • Alle COPD Patienten • Erwachsene mit Bronchiektasen unklarer Genese • Schlechtes Ansprechen einer Therapie auf late‐onset Asthma • Unklare Pannikulitis oder Antiproteinase‐3‐Vaskulitis • Angehörige ersten grades von AATD Patienten 09.01.2023
Diagnostik • Bluttest (AAT im Serum mit ELISA) Grenzwert 11yM od. 1,1g/l für Alpha1‐Antitrypsin Mangel Genotypen (zeitgleich CRP‐Bestimmung) • Schnelltests (Quick Screen Z Schnelltest, Alpha Kit, AlphaID) • Spirometrie mit Bodyplethysmographie • DLCO • Thorax Röntgen,HR‐ CT Thorax (panlobuläres basales Emphysem) • CT Thorax mit Densitometrie in „Houndsfield Einheiten“(15PD) • Leberwerte, Lebersonographie inkl. Fiberscan (ARFI) • 6MWD, SGRQ 09.01.2023
Aktuelle Empfehlung zur Diagnostik des AATM:
erst Serumspiegel bestimmen, dann genetische
Testung!
Blutstropfen
DNA
Diagnose
Niedriger AAT‐ nicht
zu PCR‐Kopie
Serumspiegel erkennen Millionen von DNA‐Kopien
in einer Stunde
1,1 g/l
(110 mg/dl) Isoelektrische PCR
Bestimmung der AAT‐ Fokussierung (z.B. AlphaID®)
Foto: pixabay
Foto: pixabay
Serumspiegel im
infektfreien Intervall
(unter bestimmten Voraussetzungen
Sequenzierung möglich)
Die finale Diagnose ist immer eine Kombination zweier Testverfahren, die sich auf
unterschiedliche biologische Testverfahren (DNA‐ und Proteinebene) beziehen.1
1. Greulich T et al. Alpha‐1‐Antitrypsin‐Mangel (AATM) – Ein Expertenstatement. Pneumologie 2020; 74: 436–442.
09.01.2023Testen mit AlphaID® 09.01.2023
AATM – eine unterdiagnostizierte
Erkrankung mit Verwechslungspotenzial
Ist der AAT‐Serumspiegel reduziert ( 1,1 g/l), sollte eine genetische Untersuchung erfolgen.1
Der AlphaID® ist ein innovatives Testkit zur Diagnose der genetischen
Erkrankung Alpha‐1‐Antitrypsin‐Mangel (AATM).
Bei dieser nichtinvasiven Testmethode wird mittels Schwämmchen ein
Wangenabstrich entnommen.
Die gewonnene DNA wird mittels In‐vitro‐Diagnostik auf die 14 häufigsten
Varianten des SERPINA1‐Gens untersucht.
Das Stäbchen (OCR‐100) ist ein Medizinprodukt mit CE‐Zertifizierung und das
Testkit enthält alle Bestandteile zur Entnahme des genetischen Materials.
Ein bakteriostatisches Reagenz hemmt das Wachstum von Bakterien. Die DNA‐Probe bleibt
bis zu 2 Monate lang stabil. Das Testkit ist bis zu 2 Jahre haltbar.
1. Schroth S et al. Alpha‐1‐Antitrypsin‐Mangel: Diagnose und Therapie der pulmonalen Erkrankung. Pneumologie 2009; 63: 335– 345.
09.01.2023AlphaID®– per Wangenabstrich zur Diagnose des AATM
Die Testung erfolgt nichtinvasiv – es ist keine Blutabnahme erforderlich!
Es wird ein einfacher Wangenabstrich durchgeführt, über den Epithelzellen
der Wangenschleimhaut entnommen werden.
Es handelt sich dabei um eine etablierte Methode aus der Kriminologie und
von Gentests zur Abschätzung von Krankheitsrisiken.
Die Analyse der Proben erfolgt im Alpha‐1‐Antitrypsin‐Zentrum Marburg.
Alpha‐1‐Antitrypsin‐Zentrum
Leitung: Prof. Dr. Timm Greulich, Prof. Dr. Claus Vogelmeier
Universitätsklinikum Gießen und Marburg
E‐Mail: alpha1@med.uni‐marburg.de
Der Befund ist ausschließlich vom Einsender auf www.AlphaID.at abrufbar.
09.01.2023AlphaID®– Identifizierung der 14
häufigsten Varianten des SERPINA1‐
Gens
Allel‐Variante Assoziierte Allele
1 c.187C>T PI*I
2 c.194T>C PI*M Procida
3 c.226_228delTTC PI*M Malton, PI*M Palermo, PI*M Nichinan
4 c.230C>T PI*S Iiyama
5 c.552delC PI*Q0 Granite falls
6 c.646+1G>T PI*Q0 West
7 c.721A>T PI*Q0 Bellingham
8 c.739C>T PI*F
9 c.839A>T PI*P Lowell, PI*P Duarte, PI*Q0 Cardiff, PI*Y Barcelona
10 c.863A>T PI*S
11 c.1096G>A PI*Z
12 c.1130dupT PI*Q0 Mattawa, PI*Q0 Ourèm
13 c.1158dupC PI*Q0 Clayton, PI*Q0 Saarbrücken
14 c.1178C>T PI*M Heerlen
09.01.2023Therapie
• Unspezifische Therapie entsprechend den Leitlinien für COPD‐
Therapie. Zusätzlich Schutzimpfungen ( Hepatitis, Pneumokokken,
Influenza). Vermeidung exogener Schadstoffe und Alkohol.
Atemphysiotherapie, pneumologische Rehabilitation.
• Spezifische Therapie mit Substitutionstherapie (Indikation AAT unter
35% vom Sollmittelwert, FeV1 zwischen 30 und 65% pred.,
Nikotinkarenz >3Monate, FeV1 decline > 50 ml/Jahr)
• Weitere apparative Optionen wie LTOT, LVRS, Ventilimplantation,
nächtliche Heimbeatmung, LUTX
06/2019 N. KaufmannSubstitution
• Gereinigtes AAT‐Konzentrat aus fraktioniertem humanen Plasma.
• Die zugelassenen Präparate von verschiedenen Pharmafirmen mit
Handelsnamen „PROLASTIN“ und „RESPREEZA“ werden in der Regel
1x/Woche intravenös in einer Dosis von 60mg/kg innerhalb 30
Minuten appliziert.
06/2019 N. KaufmannKasuistik
• 74 jähriger Pat. AATD homozygot ZZ, Substitution mit PROLASTIN
6g/Woche seit 1995, LTOT, antiobstruktive Therapie, pneum. Rehab.
Lungenfunktion: FeV1 1,21/s 36 % pred., DLCO 0,26 % pred., RV
171%pred., BGA: pO2 53 mmHg, pCO2 37 mmHg, AaDO2 47, mobil
relativ gute LQ, LUTX non vult, radiologische Progredienz in den
letzten Jahren.
06/2019 N. KaufmannAwareness ‐ short facts • Die Orphan Disease Alpha‐1‐Antitrypsinmangel (AATM) ist mit einer Prävalenz von 25 Betroffenen auf 100 000 Einwohner, nach der Amyloidose die häufigste unter den seltenen Erkrankungen. Vom pathophysiologischen Standpunkt aus ist die AATM eine Erkrankung der Leber, aufgrund der Symptomatik eine Lungenerkrankung. Der typische AATM Patient ist zwischen 30 und 50 Jahre alt, das Leitsymptom ist die Dyspnoe und die Diagnose erfolgt im Schnitt erst nach 8 Jahren. Als „undiagnosed disease“ sind in Europa weniger als 10 Prozent aller AATM Patienten erkannt. Ein Meilenstein der Behandlung war die Etablierung der Substitutionstherapie aus humanem Plasma. Schreitet die Erkrankung unerkannt und unbehandelt fort, rettet nur noch eine Doppellungentransplantation das Leben der Patienten. 09.01.2023
Sie können auch lesen

















































