Die Diagnose von Alzheimer-Demenz mittels Biomarkern
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
30
Die Diagnose von Alzheimer-Demenz mittels
Biomarkern
Dr. Mátyás Végh Ziele und Merkmale einer Diagnose
The Genetics Company, Schweiz Unter dem Begriff Diagnose werden im
Oliver Wildanger Allgemeinen drei Ziele zusammenge-
The Genetics Company, Schweiz fasst: Erstens soll die Diagnose in einer
Prof. Dr. Dr. h.c. Konrad Beyreuther sehr frühen Phase, idealerweise prä-
ZMBH Universität Heidelberg, Deutschland symptomatisch, abschätzen helfen,
wie gross das Risiko des Patienten ist,
krank zu werden. Zweitens soll die
Nach neuesten Schätzungen interna- Diagnose in einer frühen sympto-
tionaler Experten leiden heute etwa matischen Phase definitiv feststellen,
24,3 Millionen Menschen weltweit an ob der Patient an einer bestimmten
der Alzheimer-Demenz (Ferri, 2005). Krankheit leidet, während andere, ähn-
Die Zahl der AD-Patienten soll sich liche Krankheiten ausgeschlossen wer-
dabei im Jahr 2025 beinahe verdoppelt den können («Differentialdiagnose»).
haben (Ferri, 2005). Diese prognos- Drittens sollte es die Diagnose ermögli-
tizierte, dramatische Entwicklung chen, Aufschluss über den Verlauf einer
unterstreicht die Notwendigkeit und Krankheit zu erhalten, eine Prognose
Dringlichkeit von Ursachen-, Dia- zu stellen und eine allfällige Therapie
gnose- und Therapieforschung auf zu überwachen.
dem Gebiet der Alzheimer-Demenz.
Die Leistung eines Diagnostikums wird
In diesem Artikel soll ein Überblick v. a. an zwei Parametern gemessen:
über die Diagnose der Alzheimer- Sensitivität und Spezifität (s. Grafik 1).
Krankheit (Alzheimerschen Krankheit, Die Sensitivität ist ein Mass für die
Morbus-Alzheimer, Alzheimer-De- richtig positiv getesteten Patienten und
menz, AD) mittels Biomarkern gege- gibt den Prozentsatz der tatsächlich
ben werden. Biomarker stellen dabei richtig erfassten Patienten an. Bei einer
eine Alternative oder Ergänzung zu Sensititivität von 90% werden 90%
kognitiven und neuropsychologischen der Patienten mit dem Test erfasst. Die
Tests dar. Spezifität stellt eine Grösse für die
Anzahl der richtig erfassten Kontrollen
ASA SVV Alter und Lebensversicherung31
dar. Liegt die Spezifität also z.B. bei führen, jedoch meistens mit einherge-
80%, ist in 20% der Kontrollen fälsch- hender niedriger Spezifität. Um einen
licherweise jemand als krank diagnos- neuen Biomarker zu validieren muss er
tiziert worden. Die Diagnosemethode, gegen die, zu diesem Zeitpunkt, genau-
hier der gewählte Biomarker, sollte este Diagnosemethode, den «Gold-
möglichst direkt mit Ursache und standard», verglichen werden. Bei der
Pathologie in Verbindung stehen. Je Betrachtung der aus dem Vergleich er-
grösser dieser Bezug ist, desto eher mittelten Sensitivitäten und Spezifi-
kann man eine hohe Sensitivität und täten, gilt es zu beachten, dass die
Spezifität der Diagnosemethode ver- Vergleichsdiagnose, also der vermeint-
muten. Beruht die Diagnosemethode liche «Goldstandard», nicht immer die
auf Messungen von Parametern, wel- genaueste Methode ist. Dies ist insbe-
che nicht krankheitsspezifisch sind, sondere bei der Alzheimer-Demenz der
sondern nur die Begleiterscheinungen Fall. Während bei Alzheimer-Patienten
beschreiben, so kann dies unter Um- auch heute noch eine endgültige Dia-
ständen zu einer hohen Sensitivität gnose nur mittels Autopsie erstellt wer-
Grafik 1 Diagnose der Krankheit nach neuen Tests
+ –
Wahr Positiv
Diagnose + Wahr Positiv Falsch Negativ 왘 Sensitivität = Wahr Positiv + Falsch Negativ
der Krankheit
nach
Wahr Negativ
Goldstandard – Falsch Positiv Wahr Negativ 왘 Spezifität =
Wahr Negativ + Falsch Positiv
Beispiel: Ein neuer Test erreicht eine Sensitivität von 90% und eine Spezifität von 70%.
• Der Test findet 90% aller kranken Patienten, d. h. 10% der kranken Patienten werden
fälschlicherweise als gesund klassifiziert.
• Der Test findet 70% aller gesunden Patienten, d.h. 30% der gesunden Patienten werden
fälschlicherweise als krank diagnostiziert.
ASA SVV Alter und Lebensversicherung32
den kann, wird nur selten die post- nalflüssigkeit, CSF) oder technisch auf-
mortem Analyse als Vergleichsmethode wendige bildgebende Verfahren einen
hinzugezogen. Meistens dienen die mehr oder weniger direkten Einblick
weniger genauen dafür aber einfacher in die pathologischen Vorgänge der
durchführbaren kognitiven bzw. neuro- Krankheit. Weitere Schwierigkeiten bei
psychologischen Tests als designierte der Diagnose von AD sind das häufige
«Goldstandards». Für die Betrachtung Vorkommen komorbider Erkrankungen,
der hier vorgestellten Biomarker ist wie z. B. Depression und Entzündungen
dies von Bedeutung, da Studien gezeigt sowie die Koexistenz bzw. das Auf-
haben, dass kognitive bzw. neuropsy- treten von Mischformen von Demenzen,
chologische Tests, selbst wenn sie von z.B. Vaskuläre Demenz und AD.
erfahrenen Personen durchgeführt wer-
den, allenfalls nur Genauigkeiten von In den letzten Jahren hat zudem
90% erreichen (Growdon, 1999). die Patientengruppe der so genannten
leicht kognitiv gestörten Patienten
Die im Folgenden betrachteten Bio- (MCI, Mild Cognitive Impaired) an
marker weisen alle eine Sensitivität Bedeutung gewonnen. MCI-Patienten
bzw. Spezifität in einem Bereich von zeigen einzelne kognitive Störungen,
zirka 80 bis 95% auf. sind aber in ihrem täglichen Leben
wenig beeinträchtigt. Es wird heute an-
Herausforderungen an die Alzheimer- genommen, dass MCI ein Risikofaktor
Diagnose für AD ist. Pro Jahr entwickeln zirka
Die Alzheimer-Krankheit ist eine neuro- 15% der MCI-Patienten eine AD, was
degenerative Erkrankung. Das grösste über einen Zeitraum von sechs Jahren
Hindernis der Alzheimer-Demenz-Dia- in einer Konversionsrate von 80%
gnose ist somit die schlechte Zu- resultiert. Während also die meisten
gänglichkeit des betroffenen Organs, MCI-Patienten in diesem Zeitraum zur
des Gehirns. Wie später noch ausführ- AD konvertieren, entwickelt ein Teil
licher beschrieben wird, erlauben andere Demenzen und nur ein sehr klei-
Methoden wie die Analyse von Gehirn- ner Teil erfährt eine Genesung von der
Rückenmarks-Flüssigkeit (Cerebrospi- Krankheit. Das Hauptaugenmerk der
ASA SVV Alter und Lebensversicherung33
AD-Frühdiagnose ist daher zur Zeit die chemischen Markern und den Möglich-
Erkennung solcher MCI-Patienten, wel- keiten zur Diagnose von AD soll im
che eine AD entwickeln werden. Folgenden auf die für die Krankheit
charakteristischen extrazellulären und
Pathologie der Alzheimer-Demenz intrazellulären Ablagerungsstrukturen
Anfang des 20. Jahrhunderts erstellte eingegangen werden.
der deutsche Psychiater und Neuro-
pathologe Alois Alzheimer einen ersten Senile Plaques
histopathologischen Befund der später Senile oder neuritische Plaques sind
nach ihm benannten Krankheit. Erst unter physiologischen Bedingungen
etwa 80 Jahre danach wurden die von unlösliche oder nur schwer lösliche,
Alzheimer beschriebenen krankheits- dichte Proteinablagerungen von 10
typischen Proteinablagerungen, die so bis 150 Mikrometer Durchmesser. Die
genannten senilen Plaques und neuro- Hauptbestandteile dieser Aggregate
fibrillären Bündel (NFT für engl. sind verschieden lange Amyloidpeptide
Neurofibrillary Tangles) erstmals auf (Amyloid beta, Abeta oder auch Aβ).
Protein- und Genebene identifiziert und Die Länge der Abeta-Peptide variiert
charakterisiert. Die molekulare Analyse zwischen 38 und 43 Aminosäuren.
der Aggregatbestandteile führte im Neben einer Vielzahl weiterer Proteine
Folgenden zur Entwicklung diverser sind ebenfalls Astrozyten und Mikroglia
Mess- und Darstellungsmethoden, von mit den Plaques assoziiert. Dieser
denen die meisten auf Antikörper be- Umstand lässt darauf schliessen, dass
dingter Detektion basieren, sowie zur mit der Bildung der Plaques eine immu-
Entdeckung genetischer Merkmale des nologische Reaktion verbunden ist. Aβ
Krankheitsverlaufes. Somit waren die selber resultiert aus der enzymatischen
Grundlagen für die Entwicklung von Spaltung eines grösseren Vorläufer-
auf Biomarkern basierenden Diagnose- proteins APP (= Amyloid Precursor
methoden mit engem Bezug zur Protein). APP wird durch zwei enzyma-
Pathologie der Alzheimer-Krankheit tische Scheren (Sekretasen), der beta-
geschaffen. Zum besseren Verständnis Sekretase (auch BACE für engl. Beta
der Zusammenhänge zwischen bio- Amyloid Converting Enzyme) und der
ASA SVV Alter und Lebensversicherung34
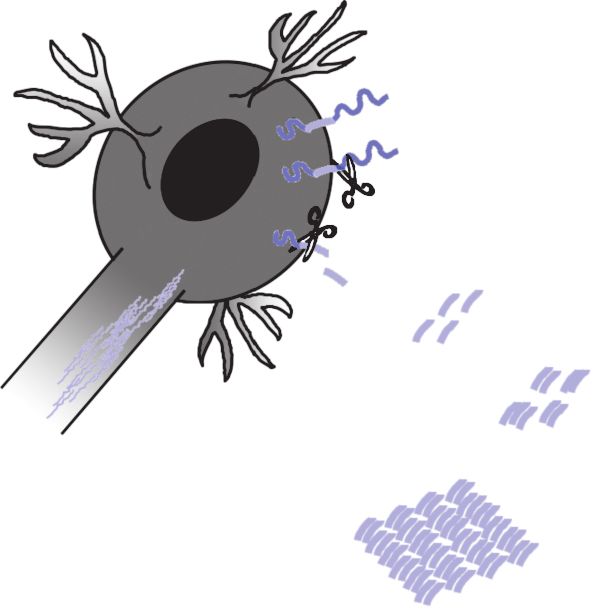
gamma-Sekretase, aufeinander fol- 왘 Grafik 2: Entstehung der Neuropathologie
in der Alzheimer-Krankheit
gend geschnitten (siehe Grafik 2). Da
Abeta (Aβ) entsteht als Schnittprodukt aus dem mem-
die gamma-Sekretase, ein Presinilin brangebundenen Vorläuferprotein APP. Für den Schnitt
enthaltender Proteinkomplex, inner- verantwortlich sind zwei ebenfalls in der Zellmembran
lokalisierte Enzyme, die beta- und die gamma-Sekretase.
halb der Membran schneidet und Mem- Einmal produziertes Abeta-Peptid aggregiert dabei
rasch zu Oligomeren und weiter zu grossen, mikrosko-
branen unterschiedlicher Dicke vor- pisch sichtbaren, unlöslichen und für die Alzheimer-
kommen, resultiert eine «Ungenauig- Krankheit typischen Ablagerungen, den senilen Plaques.
Neben der Abeta-Pathologie, findet man in den Zellen
keit» bei der Spaltung. Es entstehen so genannte neurofibrilläre Bündel aus hochgradig
phosphoryliertem Tau-Protein. Es wird heute angenom-
Abeta-Peptide unterschiedlicher Länge. men, dass bei der Alzheimer-Krankheit die Abeta-
Pathologie die Tau-Pathologie induziert. Während die
Das mit Abstand häufigste Produkt Aggregation von Tau zu einer Destabilisierung der
dieser enzymatischen Spaltung ist, mit Mikrotubuli und somit zu einer axonalen Degeneration
führt, ist die Abeta-Pathologie assoziert mit oxidativen
Anteil von nahezu 90% Abeta40, ein Stress und einer Exzitotoxizität. Die Plaques selber sind
umgeben von Microglia und Astrocyten, was zudem
Abeta-Peptid mit 40 Aminosäuren also. auf eine immunologische Reaktion schliessen lässt.
Diese Phänomene führen zu einem Verlust an Synapsen
Nur zwei Aminosäuren mehr hat das, und zum Absterben neuronaler Zellen und somit
mit einem Anteil von etwa 10%, zweit- schliesslich zur Neurodegeneration.
häufigste Peptid Abeta42. Mit Hilfe der
Massenspektrometrie konnten noch Abeta42, an das sich dann auch
weitere Formen von Abeta identifiziert Abeta40 anlagern kann.
werden (Lewzcuk, 2004b), welche aber
bislang nur wenig erforscht sind. Ob- Neben den senilen Plaques findet man,
wohl sich die beiden Hauptbestandteile auch in gesunden Menschen, so ge-
der neuritischen Plaques nur an einem nannte diffuse Plaques als extrazellu-
Ende durch zwei Aminosäuren unter- läre Amyloidablagerungen. Diese dif-
scheiden, zeigen Abeta40 und Abeta42 fusen Plaques bestehen ebenfalls aus
sehr unterschiedliche physikalische Abeta42, sind aber im Vergleich zu den
und physiologische Eigenschaften. senilen Plaques löslicher und ihre
Während Abeta40 gut löslich ist und Entstehung scheint nicht unbedingt mit
kaum aggregiert, ist Abeta42 sehr einer einhergehenden Immunreaktion
«klebrig» und neigt zur Aggregation. verbunden zu sein. Zwischenformen
Folgerichtig startet die Bildung neu- der diffusen und der senilen Plaques
ritischer Plaques vornehmlich mit bei Alzheimer-Patienten lassen ver-
ASA SVV Alter und Lebensversicherung35
Grafik 2
APP
Aβ-Produktion
β-Sekretase
γ-Sekretase
Aβ
Aβ-Oligomerisierung
Bi
ld
un
g
Ne
ur
of
ib
ril
lä
Aβ-Plaquebildung
re
Destabilisierung
rB
der Mikrotubuli
ün
de
l
Verlust von
axonalem Transport Exzitotoxizität Oxidativer Stress
왔
Aktivierung von
Microglia & Astrozyten
Neurodegeneration
Verlust von Synapsen
Neuronaler Zelltod
ASA SVV Alter und Lebensversicherung36
muten, dass diffuse Plaques Vorläufer allem aus aggregierten Tau-Proteinen.
der senilen Plaques darstellen. Tau-Proteine sind ursprünglich, durch
die Bindung an Zytoskelett-Proteine
Obwohl Nervenfasern, die in senile (sog. Mikrotubuli), für die Stabilität der
Plaques münden, Schädigungen zei- axonalen «Sendearme» der Nerven-
gen, ist die schädliche Wirkungsweise zellen mitverantwortlich. Bei der Bil-
der senilen Plaques bislang unklar. dung der fibrillären Stukturen sind
Neuere Studien machen indes deutlich, vor allem diejenigen Tau-Proteine aus-
dass senile Plaques inerte Aggregate schlaggebend, die als mehrfach phos-
eines an sich neurotoxischen Abeta- phorylierte Formen vorliegen. Aus noch
Peptides darstellen. Hinweise darauf ungeklärten Gründen wird Tau durch
lieferten Zellkulturversuche, die zeigten verschiedene Kinasen an den Binde-
das Abeta im oligomeren nicht aber stellen für Mikrotubuli phosphoryliert,
im monomeren und polymeren Zustand löst sich von den Mikrotubuli ab und
neurotoxisch ist (Kirkitadze, 2002; verklumpt zu fibrillären Strukturen in
Lesne, 2006). Die Bildung der Plaques den Neuronen. Als Folge davon ver-
als Peptidaggregate, könnte somit als lieren die Mikrotubuli an Integrität,
natürliche Massnahme zur Beseitigung so dass der axonale Transport in den
der toxischen Abeta-Peptide erklärbar Neuronen gestört und die Zellstabilität
sein. Allerdings wird vermutet, dass nicht mehr vollumfänglich gewährleis-
Plaques toxische Abeta-Peptide wieder tet ist. Die Nervenzellen sterben darauf-
freisetzen können und deshalb auch hin ab, die neurofibrillären Bündel
bei einer wirksamen Therapie entfernt werden freigesetzt. Folglich kann phos-
werden sollten. phoryliertes Tau auch extrazellulär
nachgewiesen werden. Lange Zeit war
Neurofibrilläre Bündel nicht klar, wie bzw. ob die verschiede-
Neben den extrazellulären Abeta-Abla- nen Aggregatstrukturen, die senilen
gerungen werden auch intrazelluläre Plaques sowie die neurofibrillären
Aggregatstrukturen für die Entwicklung Bündel, miteinander in Verbindung ste-
von AD verantwortlich gemacht. Diese hen. Obwohl auch heute noch nicht
neurofibrillären Bündel bestehen vor alle Fragen geklärt sind, geben ver-
ASA SVV Alter und Lebensversicherung37
schiedene Studien Hinweise darauf, altersbedingten Faktoren die lebens-
dass die neurofibrillären Bündel wo- lange Akkumulation schädigender Ein-
möglich eine Folge des Abeta-Meta- flüsse und deren Konsequenzen ge-
bolismus sind. So wurde z.B. in Maus- meint sind (Hofman, 2006).
modellen gezeigt, dass eine Gehirn-
injektion von synthetischem Abeta die Genetische Faktoren
Bildung einer Tau-Pathologie beschleu- Erst Rückschlüsse auf genetisch be-
nigt (Götz, 2001). dingte AD-Krankheitsfaktoren lieferten
Untersuchungen an Patienten mit der
Veränderung neuronaler Funktionen sogenannten familiären also vererb-
Neben den beschriebenen histologi- baren Alzheimer-Demenz (FAD) und an
schen Befunden, findet man bei einem Down Syndrom (DS) Patienten. Bei
AD-Patienten auch Veränderungen neu- FAD-Patienten können erste Symptome
ronaler Funktionen. Hier ist vor allem der AD schon relativ früh, d.h. in der
ein verminderter Glukosemetabolis- Regel in der 6. Lebensdekade, auftre-
mus, der zum eingeschränkten Ener- ten. Oftmals lassen sich erste An-
gieumsatz führt, eine verringerte zeichen einer Demenz auch schon vor
Synapsendichte und schliesslich der dem 50. Lebensjahr erkennen. Die
Verlust neuronaler Zellen zu beachten. Umstände, dass FAD-Patienten häufig
Verwandte ersten Grades mit AD vor-
Ursachen der Alzheimer-Demenz weisen, lässt auf eine genetische Ver-
Trotz des scheinbar einfachen und anlagung der Krankheit schliessen. Ein
eindeutigen pathologischen Befundes, wichtiger Hinweise für solch genetische
wird heute vermutet, dass es sich bei Veranlagungen kam auch von der Un-
der Alzheimer-Demenz eher um ein tersuchung an Down-Syndrom Patien-
multifaktorielles Syndrom, denn um ten. Bei DS-Patienten ist das 21. Chro-
eine homogene Krankheit handelt. mosom in dreifacher Ausführung vor-
Nach dem heutigen Wissensstand wer- handen (Trisomie 21). Alle Gene die
den vor allem genetische und alters- sich auf diesem Chromosom befinden,
bedingte Faktoren für den Ausbruch sind somit drei- statt zweifach vorhan-
verantwortlich gemacht. Wobei mit den und daher überdosiert. DS-Patien-
ASA SVV Alter und Lebensversicherung38
ten entwickeln, ähnlich wie FAD-Patien- schliesslich gezeigt werden, dass sol-
ten, schon sehr früh Demenzsymptome. che Mutationen zu einer erhöhten
Dies liess zunächst nur die Vermutung Abeta-Peptidproduktion und zwar
zu, dass sich auf Chromosom 21 Gene hauptsächlich des Abeta 42 führen.
befinden könnten, welche die Krank- Inzwischen konnten weitere autosomal
heitsentwicklung von AD beeinflussen. dominant vererbte Mutationen kartiert
Später konnte dasjenige Gen auf und die entsprechenden Gene, Pre-
Chromosom 21 lokalisiert werden, sinilin-1 und -2, identifiziert werden.
welches für das so genannte Amyloid Mutationen im Presinilin-1 Gen sind die
Precursor Protein (APP), also das Vor- häufigste Ursache der familiären AD.
läuferprotein der Abeta-Peptide, ko- Presiniline sind, als Teil des gamma-
diert. Die Überdosierung des APP- Sekretase-Komplexes, Enzyme, die bei
Genes führt offenbar zu einer gesteiger- der Freisetzung von Abeta aus APP die
ten Synthese der beta-Amyloidpeptide. entscheidende Rolle spielen (siehe
Tatsächlich konnten in Hirnen von Grafik 2). Sowohl die identifizierten
jungen DS-Patienten (< 20 Jahre) erste Mutationen in Presinilin-1 als auch die-
Anzeichen einer beta-Amyloidaggrega- jenigen in Presinilin-2 sind so genannte
tion beobachtet werden. Dementspre- «missense» Mutationen, welche bei der
chend entwickeln diese Patienten eine Preoteinsynthese zu einem Austausch
Demenz im prä-senilen Alter, in der von Aminosäuren führen. Im Fall von
Regel zwischen dem 50. und dem 60. Presinilin-1 und -2 bewirken diese
Lebensjahr. Änderungen in der Primärstruktur der
Enzyme eine Effizienzsteigerung und
Auch bei Patienten mit der vererbba- somit ebenfalls eine vermehrte Bildung
ren Form der Alzheimer-Demenz (FAD) von Abeta42-Peptiden.
konnten Mutationen im APP-Gen ge-
funden werden. Die gefundenen Mu- Im Gegensatz zu FAD assozierten
tationen verändern vor allem diejeni- Mutationen, welche zu Steigerungen
gen Sequenzen von APP, in denen die der Abeta42-Produktion führen, konnte
Sekretasen das Protein zur Bildung von bislang noch keine Genmutation bei
Abeta spalten. In Versuchen konnte FAD-Patienten nachgewiesen werden,
ASA SVV Alter und Lebensversicherung39
welche mit der Tau-Pathologie direkt ApoE kommt in drei verschiedenen
korrelieren. Im Gegenteil, Patienten mit Allelen (Genvarianten) vor, die als
bestimmten Mutationen im Tau-Gen, Epsilon2-, -3- und -4-Varianten bezeich-
welches auf Chromosom 17 lokalisiert net werden. Mehrere Studien zeigen,
ist, entwickeln eine so genannte fronto- dass das Epsilon4-Allel bei sporadi-
temporale Demenz mit Parkinsonismus scher AD überproportional häufig ver-
und Kopplung an Chromosom 17 (FTDP- treten ist und etwa drei Mal häufiger
17, engl. frontotemporal dementia with gefunden wird als bei der Normal-
parkinsonism linked to chromosome bevölkerung (40 – 50 Prozent gegen-
17). In post-mortem-Gehirnen von Pa- über 15 Prozent). Epsilon4 wird daher
tienten mit frontotemperaler Demenz, allgemein als Risikofaktor für die spo-
die zu den Tauopathien gerechnet wird, radische Alzheimer-Demenz betrach-
sind keine Amyloide Plaques, sondern tet. Zudem wird angenommen, dass
nur zahlreiche Neurofibrillenbündel das ApoE-Epsilon4-Allel auch beim
nachweisbar. Abeta-Metabolismus eine Rolle spielt.
Im Gegensatz zu den anderen ApoE-
Wie verschiedene wissenschaftliche Allelen werden durch das Epsilon4-Allel
Studien ergeben haben, beruhen die Abeta-Aggregate stabilisiert und da-
meisten AD-Fälle jedoch nicht auf der durch die Klärung des Abetas über die
vererbbaren sondern auf einer sporadi- CSF behindert. Ableitbar sind diese
schen Erkrankung. Inwieweit die Er- Zusammenhänge beispielsweise aus
kenntnisse der FAD Genmutationen auf der Tatsache, dass APP-transgene
eine sporadische AD übertragbar sind, Mäuse Plaques entwickeln, wenn sie
ist bislang offen. Zwar konnten einige mit ApoE-Epsilon4-Allele tragenden
Genloci identifiziert werden, die wohl transgenen Mäusen gekreuzt werden,
mit der sporadischen Erkrankung zu- während ApoE-knock-out-Mäuse kaum
sammenhängen. Die Bedeutung und Plaques entwickeln (Bales 1997).
insbesondere die Spezifität dieser
Genmutationen sind aber noch wenig Nicht-genetische Risikofaktoren
erforscht. Der wohl am besten unter- Neben den diskutierten genetischen
suchte Genlocus ist das ApoE-Gen. Risikofaktoren ist das Hauptrisiko an
ASA SVV Alter und Lebensversicherung40
einer Alzheimer-Demenz zu erkranken, bekannt. Wie schon angesprochen, ist
das Alter. Zudem ist die Prävalenzrate für die sporadische Alzheimer-Demenz
bei Frauen höher als bei Männer, nur ein besonders häufiges Vorkom-
was vielleicht auch mit der höheren men des Epsilon4-Allels des ApoE-
Lebenserwartung bei Frauen zusam- Genes beobachtet worden. Epsilon4
menhängt. Die tatsächliche Diskrepanz wird allerdings auch bei rund 15 Pro-
bezüglich der weltweiten Verteilung von zent aller gesunden Patienten gefun-
AD zwischen Entwicklungs- und Indu- den und umgekehrt sind etwa 40 bis
strieländern scheint ebenso auf dem 50 Prozent aller AD-Patienten keine
Zusammenhang von höherer Lebens- Träger des Epsilon4-Allels (Mayeux,
erwartung und höherer Prävalenzrate 1999). ApoE-Epsilon4-Träger oder nicht
zu beruhen. Weitere Faktoren, wie kann daher nicht als Ausschlusskri-
Bildung, Rauchen, vorangegangenes terium dienen und nur als Hinweis für
Schädel-Hirn-Trauma oder Aluminium- ein möglicherweise erhöhtes Risiko be-
Exposition werden zwar immer wieder trachtet werden. Zusammenfassend gilt
diskutiert, lassen aber bislang keine ein- demnach: es gibt zur Zeit keine vali-
heitlichen Schlüsse zu (Hofman, 2006). dierten genetischen Marker für die spo-
radische AD. Noch ist auch unbekannt,
Biomarker für Alzheimer-Demenz ob spontane Mutationen ebenfalls zur
Entstehung von AD beitragen können,
Genetische Marker wie dies z. B. bei Krebserkrankungen
Genetische Marker haben nicht nur das der Fall ist. Die Erforschung von geneti-
Potenzial zur Frühdiagnose sondern schen Ursachen und somit genetischen
könnten auch, da keine ethischen Be- Biomarkern für AD beschränkt sich
denken bestehen, kostengünstig und nicht auf zahllose akademische Insti-
routinemässig bei prä-symptomati- tutionen, sondern wird auch in zahl-
schen Patienten angewendet werden, reichen Firmen mit hohen Investitionen
sobald es eine wirksame Therapie gibt. betrieben. Letzteres auf Grund der
Leider sind, wie schon einleitend er- möglichen Potenziale als Diagnostika
wähnt, solche genetischen Marker nur und zur Identifizierung und Validierung
für Angehörige der FAD-Risikogruppe neuer Therapieansätze.
ASA SVV Alter und Lebensversicherung41
Biochemische Marker logischen Befunden bei AD-Patienten.
Bei der Diagnose mittels biochemischer Wie schon erwähnt, bringt das Messen
Marker werden solche Proteine, Pep- hirnspezifischer Marker ein substan-
tide oder andere chemische Stoff- zielles Problem mit sich. Das Gehirn
wechselprodukte gemessen, deren eines Patienten ist vor seinem Tod nur
Konzentrationen in Geweben oder mit sehr hohem Aufwand zugänglich.
Körperflüssigkeiten im Krankheitsfall Eine Alternative stellt, wie bereits er-
verändert sind. Für die quantitative wähnt, das Messen von Biomarkern im
Bestimmung solcher biochemischer Liquor, der Gehirn-Rückenmarks-Flüs-
Marker gibt es kostengünstige, leicht sigkeit (Cerebrospinalflüssigkeit, CSF)
skalierbare Methoden, die mit relativ dar. CSF wird dabei im Plexus chorioidei
geringem Arbeitsaufwand durchgeführt produziert und umgibt sowohl das Ge-
werden können oder sogar automati- hirn als auch das Rückenmark. Durch
sierbar sind. Im Gegensatz zu gene- den direkten Kontakt mit den Hirnzellen
tischen Markern korrelieren die Ver- wird die Zusammensetzung von CSF
änderungen biochemischer Marker von den pathologischen Vorgängen im
zeitlich und meistens sogar örtlich. Gehirn beeinflusst. In gewissem Sinne
Wobei letzteres nur gültig ist, solange ist es eine Erweiterung des neuroextra-
der Marker nicht durch Flüssigkeits- zellulären Raumes. CSF kann durch
zirkulationen im Körper verschleppt eine so genannte Lumbalpunktion ver-
wird. Häufig kann auch ein Zusammen- gleichsweise sicher entnommen wer-
hang zwischen der Markerkonzentra- den. Lumbalpunktionen sind mit ge-
tion und der Entwicklung der Krankheit ringerem Risiko behaftet als z. B.
festgestellt werden. Da die AD-Patho- Darmspiegelungen. Folglich sind in den
logie auf das Gehirn beschränkt ist, letzten Jahren mehrere CSF-Biomarker
kann davon ausgegangen werden, dass in zahlreichen Studien getestet worden
biochemische Marker möglichst hirn- (Blennow, 2004). Wie erwähnt, zeigen
spezifisch sind. Tatsächlich stehen die vor allem solche Biomarker, die direkt
zur Zeit am besten validierten Bio- mit den beiden AD-Histopathologien
marker, Tau und Abeta, im direkten Abeta und Tau im Zusammenhang ste-
Zusammenhang mit den neuropatho- hen, viel versprechende Ergebnisse:
ASA SVV Alter und Lebensversicherung42
CSF-Abeta42 heiten, z. B. Depression oder Parkinson.
Die enge Korrelation mit der Entwick- Zudem zeigen neuere Studien vermehrt
lung der Alzheimer-Demenz macht auch das diagnostische Potenzial von
das Abeta-Peptid zu einem viel ver- Abeta42 zur Früherkennung von AD bei
sprechenden Kandidaten für einen AD- MCI-Patienten (Hansson, 2006).
Biomarker. Senile Plaques bestehen
v. a. aus Abeta42. Entsprechend wer- CSF-Abeta42/40-Ratio
den post-mortem im Gehirn auch hohe Es wurde bereits darauf hingewiesen,
Abeta42-Werte gefunden, die zudem dass CSF-Abeta zu etwa 10 Prozent aus
mit der Entwicklungsstufe der Krank- Abeta42 und zu zirka 90 Prozent aus
heit korrelieren. Erstaunlicherweise Abeta40 besteht. Im Gegensatz zu CSF-
sind bei einem AD-Patienten die Werte Abeta42 scheinen CSF-Abeta40-Kon-
von löslichem (und damit im CSF mess- zentrationen nicht krankheitsspezifisch
baren) Abeta42 im CSF gegenüber Kon- zu sein. Dementsprechend verändern
trollpersonen nicht höher sondern tie- sich die CSF-Abeta40-Konzentrationen
fer. Dies steht auch im Widerspruch zu während des Krankheitsverlaufes nicht
den Werten von Abeta42 im Gehirn. Der oder nur geringfügig. CSF-Abeta40 al-
Grund für diese Diskrepanz ist bislang lein ist demnach für die AD-Diagnose
unklar. Es wird vermutet, dass bei ungeeignet. Es hat sich aber gezeigt,
fortgeschrittener Krankheit, durch die dass einzelne Patienten von Natur aus
zunehmende Plaquebildung neu gebil- höhere oder niedrigere CSF-Gesamt-
detes Abeta 42 von den Plaques ge- Abeta-Werte haben. Dabei ist die Art
bunden wird und deshalb nicht in der Abeta-Subspezies nicht von Be-
die Cerebrospinalflüssigkeit gelangt. deutung. Da Abeta40 und -42 bei sol-
Mehrere Studien haben das grosse chen Patienten gleich stark erhöht
Potenzial von Abeta42 als AD-Bio- bzw. erniedrigt sind, können Konzen-
marker beschrieben. Mit Sensitivitäten trationsvarianzen durch Einbezug von
und Spezifitäten im Bereich von rund Abeta40 als intrinsische Kontrolle
90% ist Abeta42 sehr erfolgreich bei der Gesamt-Abeta-Produktion ausge-
der Differenzierung zu anderen psy- glichen werden. Es wird also der
chiatrischen und neurologischen Krank- Verhältniswert zwischen Abeta40 zu
ASA SVV Alter und Lebensversicherung43
Abeta42 bestimmt. Erste Studien zei- hat. Bislang sind zirka 30 solcher
gen, dass eine solche Abeta42/40- Phosphorylierungsstellen beschrieben
Ratio gegenüber der Messung von worden und fast ebenso viele mögliche
Abeta42 allein eine verbesserte diag- Kinasen, welche für die Phosphory-
nostische Aussage hat. Mit der Bildung lierung von Tau verantwortlich sind
des Verhältnisses der beiden Abeta- (Bueé, 2000). Dementsprechend gross
Peptide lassen sich Sensitivitäten und ist das Marktangebot für Antikörper,
Spezifitäten von bis zu 96% erreichen die die verschiedenen Phosporylie-
(Lewczuk, 2004a; Hansson, submitted). rungsstellen an Tau selektiv erkennen.
Es hat sich jedoch gezeigt, dass für die
CSF-tTau Diagnose von AD vor allem pTau181
Im CSF erhöhtes tTau-Protein oder total (Tau phosphoryliert an der Aminosäure
Tau, also die Gesamtmenge aller Tau- der Position 181), pTau199 und
subspezies, ist ein Zeichen neuronaler pTau231 relevant sind. Als separate
Degeneration. Folglich werden tTau- Parameter zeigen die verschiedenen
Konzentrationen nicht nur als Dia- Tau-Subspezies ähnliche Sensitivitäten
gnoseparameter für die Alzheimer- und Spezifitäten (Hampel, 2004), die
Demenz, sondern auch für andere zwischen zirka 81 bzw. 91 Prozent
neurodegenerative Erkrankungen wie liegen. Im Vergleich zum tTau-Marker
z. B. der Creutzfeld-Jakob-Krankheit ge- verfügt pTau anscheinend über das
nutzt. Sensitivitäten und Spezifitäten grössere Potenzial, da einige Phos-
liegen bei diesem Parameter bei durch- phorylierungen, die in anderen Tau-
schnittlich 81 – 91%. opathien nicht vorkommen, doch eher
AD-spezifisch sind.
CSF-pTau
Neurofibrilläre Bündel bestehen haupt- Plasma-Abeta
sächlich aus hyperphosphorylierten Leider gibt es zurzeit keine einheitli-
Tau-Proteinen, also aus Tau welches chen Daten bezüglich einer Korrelation
an diversen Stellen, vornehmlich an der Konzentrationen von Plasma-Abeta
den beiden Aminosäuren Serin und und CSF-Abeta (Irizarry, 2004). Wäh-
Threonin, Phosphatgruppen gebunden rend CSF-Abeta42 mit der Entwicklung
ASA SVV Alter und Lebensversicherung44
der Alzheimer-Demenz sehr gut korre- in einer gross angelegten klinischen
liert, besteht Grund zur Annahme, dass Studie herausgefunden, dass hohe
nicht alles Abeta im Blut neurologi- Plasma-Abeta40-Werte, vor allem in
schen Ursprungs ist. Zudem stossen Kombination mit tiefen Plasma-Abe-
derzeit Versuche zur Bestimmung von ta42-Werten, ein erhöhtes Demenz-
Plasma-Abeta an technische Grenzen, risiko darstellen (van Oijen, 2006). Die
da die Gesamt-Abeta-Konzentration im Anwendbarkeit dieser Schlussfolge-
Blut etwa zehnmal geringer ist als im rung zur Risikobeurteilung eines ein-
CSF. Zudem ist die Konzentration von zelnen Patienten muss jedoch noch in
Abeta42 im Plasma, genauso wie in weiteren Studien bestätigt werden.
CSF, etwa um einen Faktor 10 tiefer ist
als die Konzentration von Abeta40. Andere biochemische Plasma-Marker
Die zur Zeit erhältlichen Messmethoden Die im vorherigen Abschnitt dargestell-
erreichen die notwendigen statistisch ten Probleme bei der zuverlässigen
absicherbaren Detektionslimits für Messung von Abeta-Peptiden in Plas-
Abeta42 somit nicht. maproben, veranlasste viele Wissen-
schafter zur Suche nach weiteren
Hinzu kommt, dass Abeta42, bedingt krankheitspezifischen Biomarkern. Zahl-
durch seine physikalischen Eigen- reiche molekulare Nebenprodukte der
schaften, mit einer Vielzahl von ver- Alzheimer-Demenz-Pathologie wurden
schiedenen Proteinen interagiert, so als Alternativen zu Abeta untersucht
z.B. mit Albumin oder Heparin, welche (Teunissen, 2003). Zu den potenziellen
natürlich in hohen Mengen im Plasma AD-Markern gehören unter anderem
vorkommen oder zu den Blutproben zu- Isoprostan, 3-Nitrotyrosin (oxidativer/
gesetzt wurden. Diese Bindung mit an- nitrativer Stress); alfa1-Antichymotryp-
deren Plasmabestandteilen verhindert sin, Interleukine (Entzündungen); C-
die genaue Bestimmung sowie die reaktives Protein, C1q (Komplement-
Reproduzierbarkeit der Daten. Neuere system); 24S-Hydroxycholesterol (spe-
Studien zeigen aber, dass zumindest zisches Produkt des Cholesterin Meta-
für Plasma-Abeta40 gewisse Trends bolismus des Gehirns) und Homo-
auszumachen sind. So wurde kürzlich cystein. Auf Grund niedriger Sensi-
ASA SVV Alter und Lebensversicherung45
tivitäten und v.a. Spezifitäten erreicht Bei den bildgebenden Verfahren wird
jedoch keiner dieser Marker das not- im Allgemeinen zwischen drei Ansätzen
wendige Potenzial eine zuverlässig unterschieden. Die Visualisierung und
Diagnose von AD zu stellen. Zudem Auswertung struktureller oder funktio-
können die Konzentrationen mancher neller Veränderungen des Gehirns so-
Markerkandidaten im Blutplasma, z.B. wie die Plaqueentwicklung dienen hier-
durch die Einahme von Vitaminprä- bei als Parameter der pathologischen
paraten, Begleitmedikamenten oder Entwicklung. In den folgenden Ab-
komorbiden Erkrankungen stark beein- schnitten werden diese Ansätze weiter
flusst werden. erläutert.
Bildgebende Verfahren Visualisierung struktureller
Im Gegensatz zur Messung von Bio- Veränderungen
markern im CSF und Blutplasma, bieten Strukturelles Neuroimaging dient vor
bildgebende Verfahren (engl. Neuro- allem der Entdeckung und Vermessung
imaging) die Möglichkeit die pathologi- von Atrophien des Gehirns bzw. Teilen
schen Vorgänge der Alzheimer-Demenz des Gehirns. In Verbindung mit AD ist
schon in sehr frühen Stadien direkt im vor allem die Volumenabnahme des
Gehirn zu beobachten. Hohe Anschaf- Hippokampus zu nennen. Zur Visua-
fungs-, Betriebs- sowie Messkosten lisierung derartiger Strukturen werden
verhindern jedoch zur Zeit noch die sowohl Computertomographen (CT) als
schnelle Verbreitung dieser Methode auch immer häufiger Magnetresonanz-
in der präklininischen und klinischen Tomographen (MRT) benutzt. Neben
Forschung sowie in der klinischen oder der sensitiven Diagnose von Alzheimer-
ambulanten Diagnostik. An der Weiter- Demenz, erlauben diese Verfahren
entwicklung dieser Methode wird da- auch die Erforschung und Differenzie-
her umso intensiver in vielen akade- rung anderer Ursachen kognitiver Stö-
mischen und industriellen Institutionen rungen wie zum Beispiel Wasserkopf
gearbeitet. (Hydrocephalus) oder Gehirntumore
(z.B. Glioblastome). Das Auflösungsver-
mögen heutiger MRT-Anlagen ist dabei
ASA SVV Alter und Lebensversicherung46
schon so gross, dass selbst kleinste sions-Tomographie (PET) bildlich dar-
strukturelle Veränderungen nachge- gestellt. SPECT ermöglicht die Visua-
wiesen werden können. Dennoch eig- lisierung der Blutzirkulation im Gehirn
net sich die Visualisierung struktureller durch Gammastrahlen detektierende
Veränderungen des Gehirns generell Kameras. Als Quelle der Gamma-
nur für die post-symptomatische Dia- strahlen dient 99mTc markiertes Hexa-
gnose, da ein gewisser Degenerations- methylpropylen-Amin-Oxim (HMPAO),
grad von neuronalem Gewebe erreicht ein fettlösliches Radionuklid, das dem
werden muss, um eine zuverlässige Patienten vor der Untersuchung inji-
Diagnose stellen zu können. Werden ziert wird. Die Auswertung der Bilder
jedoch serielle Messungen vorgenom- ergibt eine semiquantitative Analyse
men um die Degenerationsrate zu be- des Blutdurchflusses. Gegenüber Kon-
stimmen, kann die Magnetresonanz- trollpersonen, weisen AD-Patienten vor
Tomographie schon früh zu einer zuver- allem in den tempoparietalen Regionen
lässigen Diagnose beitragen. Serielle einen verminderten Blutdurchfluss auf.
Messungen sind zudem zur Evaluierung Trotz relativ guter diagnostischer Dif-
von möglichen neurodegenerationsver- ferenzierung zwischen AD und ande-
zögernden oder -stoppenden Therapien ren Demenzformen, zeigt sich bislang
geeignet. nur wenig klinisch valider Nutzen des
SPECT-Verfahrens zur AD-Früherken-
Funktionelle Veränderungen nung. Mehr als SPECT hat die 18FDG
Im Vergleich zum strukturellen Neuro- PET in verschiedenen Studien ihr
imaging wird allgemein dem so genan- Potenzial als AD-Früh- und -Differen-
ten funktionellen Neuroimaging ein zialdiagnosemethode unter Beweis ge-
noch grösseres Potenzial zur frühen stellt (Silvermann, 2001). AD-Patienten
AD-Diagnose sowie zur Differentialdia- zeigen schon früh einen verminderten
gnose zu gesprochen. Bei diesem Ver- Metabolismus in bestimmten Hirn-
fahren wird die Durchblutung oder regionen (z.B. tempoparietal). Mittels
der Stoffwechsel des Gehirns mittels 18F-markierter Fluor-2-deoxy-2-D-Glu-
Single - Photonen - Emissions -Tomogra- cose (18FDG) kann die Glucoseauf-
phie (SPECT) bzw. Positronen-Emis- nahme und damit der Energiebedarf
ASA SVV Alter und Lebensversicherung47
neuronaler Regionen bestimmt werden. und Abeta40 mit einbeziehen, sind je-
18FDG wird dabei in Zellen in FDG-6- doch geplant.
Phosphat umgewandelt, welches nicht
mehr weiter glycolytisch metabolisiert Darstellung von Plaques
wird und in den Neuronen akkumuliert. Um die Korrelation zur pathologischen
Die über einen Zeitraum von 30 bis 40 Entwicklung der Alzheimer-Demenz
Minuten akkumulierte Menge korreliert noch zu erhöhen, wurden in letzter Zeit
mit dem Glucosedurchsatz und somit PET-Tracer zur Visualisierung seniler
mit dem Energiebedarf bzw. der Akti- Plaques entwickelt. 8FFDDNP (2-(1-
vität der Zellen. (6-(2-(18F)fluoroethyl)(methyl)amino)-
2naphtyl)ethyliden)malononitrile) (Ag-
Das Diagnosepotenzial sowohl von deppa, 2001) und PIB (Pittsburgh
SPECT als auch von PET ist durch die Compound-B, ein Thioflavin-T-Derivat)
räumliche Auflösung der Methoden (Klunk, 2004) sind zwei Beispiele von
begrenzt. Die gemessenen Werte sind Substanzen, die an senile Plaques
Mittelwerte eines bestimmten Volu- binden und für das PET detektierbar
mens. Sie können durch die ungenü- machen. Erste Resultate sind sehr viel
gende Auflösung und durch die zu- versprechend, jedoch fehlen noch grös-
nehmende Atrophie des Gewebes ver- sere Studien, insbesondere auch Kor-
fälscht werden. Teilweise können ver- relationsstudien unter Einbeziehung
fälschte Werte durch simultane CT oder des CSF-Abeta42/Abeta40-Verhältnis-
MRT korrigiert werden. ses, die den diagnostischen Nutzen
dieser Methode belegen.
Obwohl einige publizierte Studien vor
allem für die 18FDG PET sehr viel ver-
sprechende Resultate zeigen, sind die-
se Methoden erst wenig standardisiert
und klinisch validiert. Mehrere umfang-
reiche Studien, die einer Standardi-
sierung und Validierung dienen können
und biologische Marker wie Abeta42
ASA SVV Alter und Lebensversicherung48
Schlussfolgerungen Angehörigen früh auf die Krankheit
Obwohl die meisten hier beschriebe- einstellen und gemeinsam allfällige
nen Methoden bislang eher zu For- Massnahmen ergreifen (Leifer, 2003).
schungszwecken eingesetzt werden,
stehen den spezialisierten Kliniken Untersuchungen und Umfragen in den
heutzutage neben neuropsychologi- USA haben aber gezeigt, dass viele
schen und kognitiven Tests eine ganze AD-Patienten zu spät, im Schnitt erst
Batterie an Prüfmethoden für eine frühe 4 Jahre nach Ausbruch der Krankheit,
Erkennung der Alzheimer-Demenz zur erkannt werden. Die Gründe dafür sind
Verfügung. vielfältig und reichen von der Stig-
matisierung der Krankheit über das
Stellt sich die Frage, warum eine frühe Negieren erster Symptome durch den
Diagnose von AD überhaupt sinnvoll Patienten bis hin zur Fehldiagnose
ist, obwohl die Krankheit bislang als durch den Arzt. Hier wird deutlich,
nicht heilbar gilt. Tatsache ist jedoch, dass Aufklärung über die Krankheit
dass etwaige Medikamente, die zur Zeit ebenso wichtig ist wie die Fortsetzung
auf dem Markt angeboten werden, ihre der Forschung an Diagnosemethoden
palliativen Wirkungen umso besser ent- und Therapieansätzen der Alzheimer-
falten, je früher die Diagnose gestellt Demenz.
und das Medikament eingenommen
wurde (Chang und Silvermann, 2004). Alle hier vorgestellten Diagnoseme-
Somit trägt die Früherkennung der thoden haben ihre Vor- und Nachteile.
Krankheit auch zu einer Verbesserung Letztere betreffen beispielsweise
der Lebensqualität des Patienten und Infektionsrisiken bei Lumbalpunktio-
häufig auch der Angehörigen bei. Die nen oder Strahlenbelastung beim
Verbesserung der Lebensqualität bei Neuroimaging. Es scheint, dass die
frühzeitiger Diagnose von AD erhöht einzelnen Tests nur selten über
zudem die Möglichkeit, bei Früherken- Sensitivitäten bzw. Spezifitäten von
nung die hohen Finanzaufwendungen 90 Prozent hinauskommen. Auf den
der Krankenpflege zu reduzieren. Zu- ersten Blick gibt es, abgesehen von den
dem können sich der Patient und die Kosten, keinen wirklichen Vorteil der
ASA SVV Alter und Lebensversicherung49
Biomarker gegenüber den kognitiven erster Schritt auf dem Weg zu einer
Tests. Das eigentliche Potenzial der personalisierten und hoffentlich wirk-
Biomarker liegt neben dem Kosten- samen und gut verträglichen Therapie
vorteil jedoch klar in der prä-sympto- eingesetzt zu werden.
matischen Früherkennung der Krank-
heit. Der Aspekt des quantitativen Referenzen
Monitorings des Krankheits- bzw. Hei- • Agdeppa ED, Kepe V, Liu J, Flores-
lungsverlaufes durch Biomarker wird Torres S, Satyamurthy N, Petric A,
bei der Zulassung erster zielgerichteter Cole GM, Small GW, Huang SC,
und somit krankheitsverändernder Me- Barrio JR. Binding characteristics of
dikamente (targeted drugs) deutlich radiofluorinated 6-dialkylamino-2-
werden. Therapieansätze, die auf die naphthylethylidene derivatives
Verhinderung der Bildung von Abeta- as positron emission tomography
Peptiden, die Entfernung von Plaques imaging probes for beta-amyloid
durch z. B. Impfung oder die Unter- plaques in Alzheimer’s disease.
bindung der Phosphorylierung von Tau J Neurosci.
fokussieren, sind Beispiele, für die 2001 Dec 15; 21(24): RC189.
quantitatives Monitoring schon heute • Bales KR, Verina T, Dodel RC, Du Y,
eingesetzt wird. Wenn auch erst in den Altstiel L, Bender M, Hyslop P,
klinischen Phasen der Medikamenten- Johnstone EM, Little SP, Cummins DJ,
entwicklung. Piccardo P, Ghetti B, Paul SM.
Lack of apolipoprotein E dramatically
Vorstellbar wäre zukünftig auch eine reduces amyloid beta-peptide
Einteilung der Demenzen basierend auf deposition. Nat Genet.
Grundlagen der Biomarkertechnologie 1997 Nov; 17(3):263 – 4.
anstatt kognitiver Tests. Diese hätte zu- • Blennow K. Cerebrospinal fluid
dem den Vorteil, dass eine Behandlung protein biomarkers for Alzheimer’s
entsprechend früher und zielgerichte- disease. NeuroRx.
ter bzw. spezifischer eingeleitet werden 2004 Apr; 1(2):213 – 25.
könnte. Wir sind überzeugt, dass Bio- • Buee L, Bussiere T, Buee-Scherrer V,
marker das Potenzial haben, als ein Delacourte A, Hof PR. Tau protein
ASA SVV Alter und Lebensversicherung50
isoforms, phosphorylation and role Moller HJ, Davies P, Blennow K.
in neurodegenerative disorders. Measurement of phosphorylated tau
Brain Res Brain Res Rev. epitopes in the differential diagnosis
2000 Aug; 33(1):95 – 130. of Alzheimer disease: a comparative
• Chang CY, Silverman DH. Accuracy cerebrospinal fluid study. Arch Gen
of early diagnosis and its impact Psychiatry.
on the management and course of 2004 Jan; 61(1):95 – 102.
Alzheimer’s disease. Expert Rev Mol • Hansson O, Zetterberg H, Buchhave
Diagn. 2004 Jan; 4(1):63 – 9. P, Londos E, Blennow K, Minthon L.
• Ferri CP, Prince M, Brayne C, Brodaty Association between CSF biomarkers
H, Fratiglioni L, Ganguli M, Hall K, and incipient Alzheimer’s disease in
Hasegawa K, Hendrie H, Huang Y, patients with mild cognitive impair-
Jorm A, Mathers C, Menezes PR, ment: a follow-up study. Lancet
Rimmer E, Scazufca M; Alzheimer’s Neurol. 2006 Mar; 5(3):228 – 34.
Disease International. Global • Hansson O., Zetterberg H., Buchhave
prevalence of dementia: a Delphi P., Andreasson U., Minthon L.,
consensus study. Lancet. Blennow K., Prediction of
2005 Dec 17; 366(9503):2112 – 7. Alzheimer’s disease using CSF
• Gotz J, Chen F, van Dorpe J, Nitsch Abeta42 and Abeta42/40 ratio in
RM. Formation of neurofibrillary patients with mild cognitive impair-
tangles in P301l tau transgenic mice ment, manuscript submitted.
induced by Abeta 42 fibrils. Science. • Hofman A., de Jong P.T.V.M.,
2001 Aug 24; 293(5534):1491 – 5. van Duijn C.M., Breteler M.M.B.,
• Growdon JH. Biomarkers of Epidemiology of neurological dis-
Alzheimer disease. Arch Neurol. eases in elderly people: what did we
1999 Mar; 56(3):281 – 3. learn from the Rotterdam study.
• Hampel H, Buerger K, Zinkowski R, Lancet Neurol.
Teipel SJ, Goernitz A, Andreasen N, 2006 May; 5:545 – 550.
Sjoegren M, DeBernardis J, Kerkman • Irizarry MC. Biomarkers of Alzheimer
D, Ishiguro K, Ohno H, Vanmechelen disease in plasma. NeuroRx.
E, Vanderstichele H, McCulloch C, 2004 Apr; 1(2):226 – 34.
ASA SVV Alter und Lebensversicherung51
• Kirkitadze MD, Bitan G, Teplow DB. Reulbach U, Kornhuber J, Wiltfang J.
Paradigm shifts in Alzheimer’s dis- Neurochemical diagnosis of
ease and other neurodegenerative Alzheimer’s dementia by CSF
disorders: the emerging role of oli- Abeta42, Abeta42/Abeta40 ratio
gomeric assemblies. J Neurosci Res. and total tau. Neurobiol Aging.
2002 Sep 1; 69(5):567 – 77. 2004 Mar; 25(3):273 – 81. (a)
• Klunk WE, Engler H, Nordberg A, • Lewczuk P, Esselmann H, Groemer
Wang Y, Blomqvist G, Holt DP, TW, Bibl M, Maler JM, Steinacker P,
Bergstrom M, Savitcheva I, Huang Otto M, Kornhuber J, Wiltfang J.
GF, Estrada S, Ausen B, Debnath ML, Amyloid beta peptides in cerebro-
Barletta J, Price JC, Sandell J, Lopresti spinal fluid as profiled with surface
BJ, Wall A, Koivisto P, Antoni G, enhanced laser desorption/
Mathis CA, Langstrom B. Imaging ionization time-of-flight mass
brain amyloid in Alzheimer’s disease spectrometry: evidence of novel
with Pittsburgh Compound-B. biomarkers in Alzheimer’s disease.
Ann Neurol. Biol Psychiatry.
2004 Mar; 55(3):306 – 19. 2004 Mar 1; 55(5):524 – 30. (b)
• Leifer BP. Early diagnosis of • Mayeux R, Saunders AM, Shea S,
Alzheimer’s disease: clinical and Mirra S, Evans D, Roses AD, Hyman
economic benefits. J Am Geriatr Soc. BT, Crain B, Tang MX, Phelps CH.
2003 May; 51(5 Suppl Dementia): Utility of the apolipoprotein E geno-
281 – 8. type in the diagnosis of Alzheimer’s
• Lesne S, Koh MT, Kotilinek L, disease. Alzheimer’s Disease Centers
Kayed R, Glabe CG, Yang A, Consortium on Apolipoprotein E and
Gallagher M, Ashe KH. A specific Alzheimer’s Disease. N Engl J Med.
amyloid-beta protein assembly in 1998 Feb 19; 338(8):506 – 11.
the brain impairs memory. Nature. • Silverman DH, Small GW, Chang CY,
2006 Mar 16; 440(7082):352 – 7. Lu CS, Kung De Aburto MA, Chen W,
• Lewczuk P, Esselmann H, Otto M, Czernin J, Rapoport SI, Pietrini P,
Maler JM, Henkel AW, Henkel MK, Alexander GE, Schapiro MB, Jagust
Eikenberg O, Antz C, Krause WR, WJ, Hoffman JM, Welsh-Bohmer KA,
ASA SVV Alter und Lebensversicherung52
Alavi A, Clark CM, Salmon E, de Leon Empfohlene Reviews
MJ, Mielke R, Cummings JL, Kowell und weiterführende Literatur
AP, Gambhir SS, Hoh CK, Phelps ME.
Positron emission tomography Allgemein
in evaluation of dementia: Regional • Alzheimer-Demenz, Hampel-
brainmetabolism and long-term Padberg-Möller, Wissenschaftliche
outcome. JAMA. Verlagsgesellschaft mbH Stuttgart,
2001 Nov 7; 286(17):2120 – 7. 2003 (nur in Deutsch erhältlich).
• Teunissen CE, Lutjohann D, von • Fokus auf Alzheimer Demenz im
Bergmann K, Verhey F, Vreeling F, Journal Nature Medicine,
Wauters A, Bosmans E, Bosma H, Ausgabe Juli 2006, Vol 12 Nr. 7:
van Boxtel MP, Maes M, Delanghe J, http://www.nature.com/nm/focus/
Blom HJ, Verbeek MM, Rieckmann P, alzheimer/index.html
De Bruijn C, Steinbusch HW, • Masters CL, Cappai R, Barnham KJ,
de Vente J. Combination of serum Villemagne VL. Molecular mechan-
markers related to several mechan- isms for Alzheimer’s disease:
isms in Alzheimer’s disease. implications for neuroimaging and
Neurobiol Aging. therapeutics. J Neurochem.
2003 Nov; 24(7):893 – 902. 2006 Jun; 97(6):1700 – 25.
• van Oijen M, Hofman A, Soares HD, • Selkoe DJ. Alzheimer’s disease:
Koudstaal PJ, Breteler MM. Plasma genes, proteins, and therapy. Physiol
Abeta(1 – 40) and Abeta(1 – 42) and Rev. 2001 Apr; 81(2):741 – 66.
the risk of dementia: a prospective
case-cohort study. Lancet Neurol. Genetik
2006 Aug; 5(8):655 – 60. • Selkoe DJ, Podlisny MB. Deciphering
the genetic basis of Alzheimer’s
disease. Annu Rev Genomics Hum
Genet. 2002; 3:67 – 99.
Epub 2002 Apr 15.
ASA SVV Alter und Lebensversicherung53
Biochemische Biomarker Bildgebende Verfahren
• Blennow K. Cerebrospinal fluid • Petrella JR, Coleman RE, Doraiswamy
protein biomarkers for Alzheimer’s PM. Neuroimaging and early
disease. NeuroRx. diagnosis of Alzheimer disease:
2004 Apr; 1(2):213 – 25. a look to the future. Radiology.
• Irizarry MC. Biomarkers of Alzheimer 2003 Feb; 226(2):315 – 36.
disease in plasma. NeuroRx. • Jagust W. Molecular neuroimaging
2004 Apr; 1(2):226 – 34. in Alzheimer’s disease. NeuroRx.
• Teunissen CE, de Vente J, Steinbusch 2004 Apr; 1(2):206 – 12.
HW, De Bruijn C. Biochemical mar-
kers related to Alzheimer’s dementia
in serum and cerebrospinal fluid. Links
Neurobiol Aging. • Alzforum: Die umfassendste
2002 Jul – Aug; 23(4):485 – 508. Internetseite sowohl für Laien als
• Teunissen CE, Lutjohann D, von auch für das Fachpublikum:
Bergmann K, Verhey F, Vreeling F, http://www.alzforum.org/
Wauters A, Bosmans E, Bosma H,
van Boxtel MP, Maes M, Delanghe J, • Öffentliche Datenbank für mit AD
Blom HJ, Verbeek MM, Rieckmann P, korrelierende Gene:
De Bruijn C, Steinbusch HW, de http://www.alzforum.org/res/com/
Vente J. Combination of serum mar- gen/alzgene/
kers related to several mechanisms
in Alzheimer’s disease. Neurobiol • Webseite der VCDN-Gruppe
Aging. (Englisch und Französisch):
2003 Nov; 24(7):893 – 902. Viel Information und grafische
Darstellungen über Tau- und Abeta-
Pathologie:
http://www.alzheimer-adna.com/
english.html
ASA SVV Alter und LebensversicherungSie können auch lesen

















































