Demenz und Neurodegeneration - WS 2020/21 Vorlesung Neuropathologie - PD Dr. Astrid Jeibmann
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
WS 2020/21 Vorlesung Neuropathologie
Demenz und Neurodegeneration
PD Dr. Astrid Jeibmann
Institut für Neuropathologie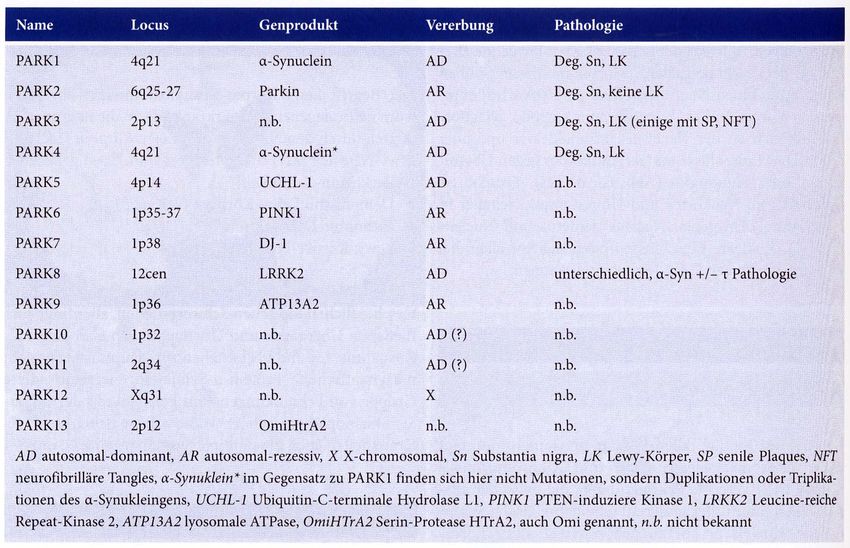
Kennen Sie...?
Warum ist das wichtig?
• Wieviel % der Menschen mit 70Lj. sind betroffen?
5%
• Wieviel % der Menschen mit 85Lj. sind betroffen?
35%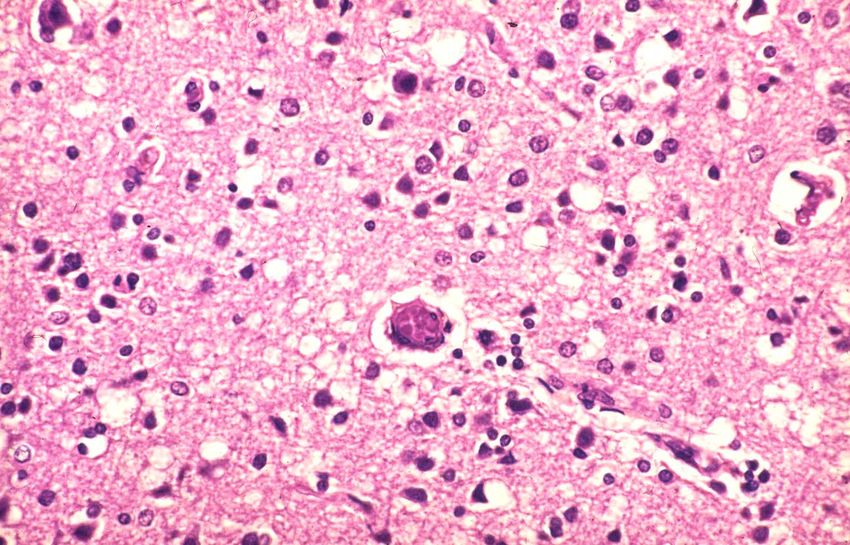
Warum ist das wichtig?
Warum ist das wichtig?
Werde ich dement?
Namsolleck et al. 2014
BMFSFJ 2015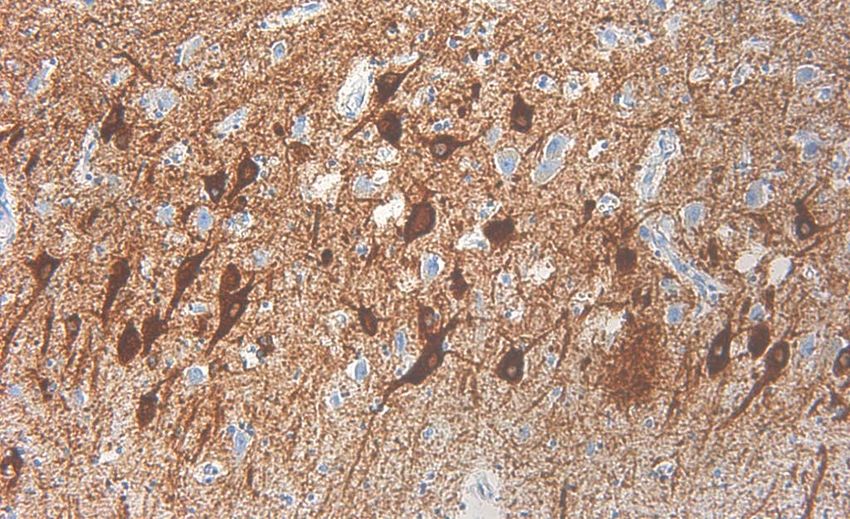
Wie fällt Demenz auf?
Wie fällt Demenz auf? • Beeinträchtigung intellektueller Fähigkeiten • Erworben • Persistierend • Keine Bewusstseinsstörung • Beeinträchtigung des Alltagslebens • Mehrere kognitive Bereiche betroffen

Welche Rolle spielt die Neuropathologie? Die definitive Artdiagnose einer Demenz ist nur durch die histopathologisch- autoptische Untersuchung möglich. Die Autopsie dementer Patienten ist notwendig: •Zur Sicherung oder Abweisung klinischer Diagnosen, •zur Validierung diagnostischer Kriterien, •für epidemiologische und therapeutische Studien und •für die genetische Beratung. Bei jedem verstorbenen dementen Patienten sollte eine autoptische Untersuchung des Gehirns unter Anwendung optimaler Methoden und standardisierter Diagnosekriterien erfolgen.
Ursachen der Demenz
Alzheimer-Krankheit Vaskuläre
Demenz
Frontotemporale
Demenz
Lewy-Körper-Demenz/
M. Parkinson
Andere
NeurodegenerationenUrsachen der Demenz
Alzheimer-Krankheit Vaskuläre
Demenz
Frontotemporale
Demenz
Lewy-Körper-Demenz/
M. Parkinson
Andere
NeurodegenerationenGeschichte Alois Alzheimer Auguste Detert
Geschichte
"Wie heißen Sie?"
"Auguste."
"Familienname?"
"Auguste."
"Wie heißt ihr Mann?" - Auguste Deter zögert, antwortet schließlich:
"Ich glaube... Auguste."
"Ihr Mann?"
"Ach so."
"Wie alt sind Sie?"
"51."
"Wo wohnen Sie?"
"Ach, Sie waren doch schon bei uns."
"Sind Sie verheiratet?"
"Ach, ich bin doch so verwirrt."
"Wo sind Sie hier?"
"Hier und überall, hier und jetzt, Sie dürfen mir nichts übel nehmen."
"Wo sind Sie hier?"
"Da werden wir noch wohnen."
"Wo ist Ihr Bett?"
"Wo soll es sein?"Geschichte
Merkwürdige Veränderungen der Neurofibrillen
Miliare Herdchen durch Einlagerung
eines eigenartigen StoffesAlzheimer Krankheit - Klinik
• >65 Jahre
• Merkleistungs- und
Wortfindungsstörungen
• Zeitliche und räumliche
Desorientierung
• Persönlichkeits-
veränderungen
• Innerhalb von 5-8 Jahre
J Neural Transm (2006) 113: 1603-1623
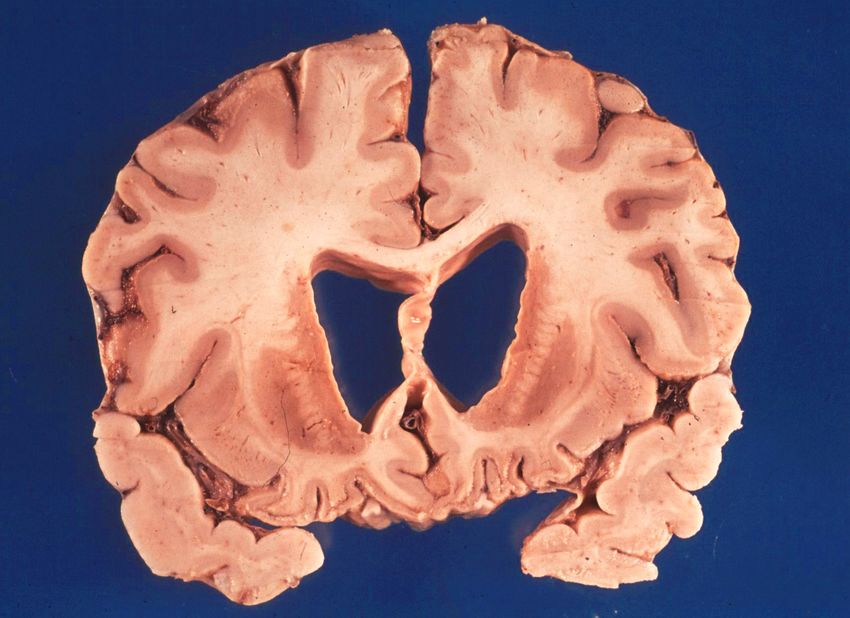
Pflegebedürftigkeit und TodMakro- und mikroskopische Befunde
gesund AD
• Hirnatrophie
– Verkleinerung des
Kortex
– Vergrößerung der
Ventrikel
verändert nach American Health Assistance Foundation
• Beta-Amyloid-
(Aβ)Plaques
• Tangles
verändert nach Acta Neuropathol (2009) 118: 5-36Makroskopie Morbus Alzheimer Normal
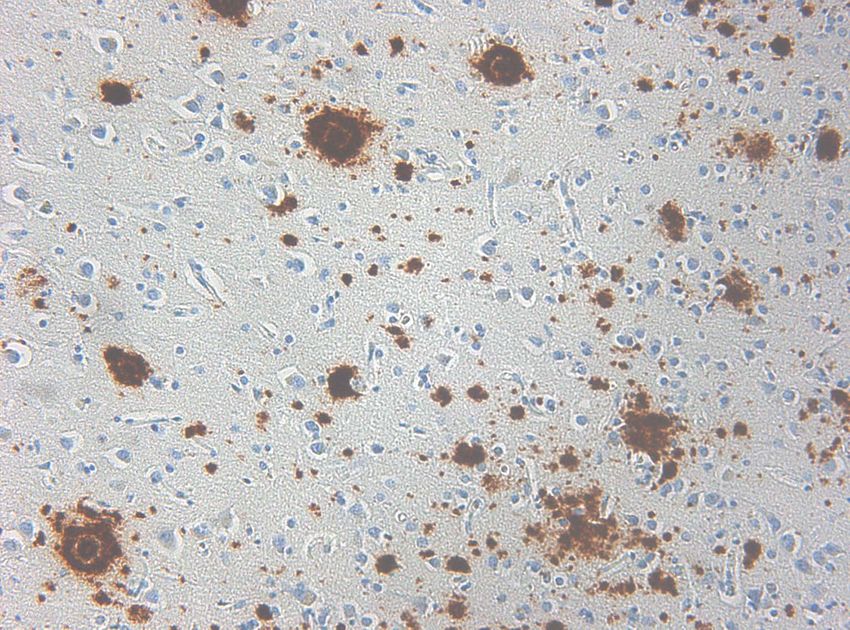
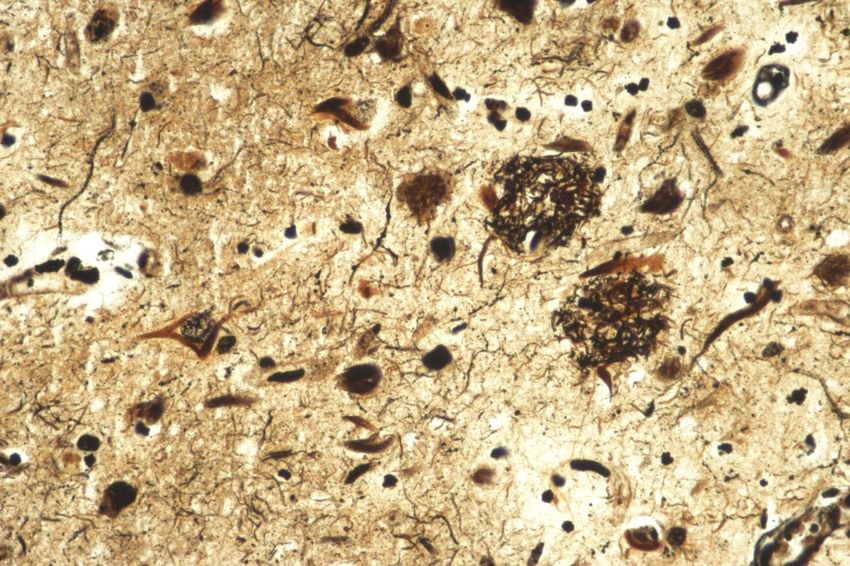
Histologie
Kernplaque
(cored Plaque)
Diffuser
Plaque
Plaques: Aß, ßA4, ß-AmyloidGenetik I
- Familiäre Form
- vor dem 65. Lebensjahr (häufig 40-65)
- 0,5% der Patienten haben autosomal-dominant vererbte
Alzheimer-Krankheit
- in der Regel (40%) durch Mutationen in APP, Präsenilin-1 oder
Präsenilin-2 verursacht „early-onset Alzheimer‘s disease“
Chromosom Gen Genprodukt
21 APP Amyloid-Vorläufer-Protein
14 PSEN1 Präsenilin-1
1 PSEN2 Präsenilin-2
- Trisomie 21! Morphol. Veränderungen ab 20.LJ, Demenz ab 35. LJGenetik II
- Sporadische Formen
- nach dem 65. Lebensjahr, über 90% der Fälle
- eine eindeutige Zuordnung zu einer Mutation ist bei dieser Form
nicht möglich – das Apolipoprotein-E-e4 Allel ist derzeit der einzige
gesicherte genetische Risikofaktor
- ein e4-Allel (heterozygot): 4-fach höheres Risiko für AD
- zwei e4-Allele (homozygot): 18-fach höheres Risiko für AD
- Bei sporadischen Fällen keine vermehrte Produktion von Aß,
möglicherweise Problem beim Abbau von AßPathomechanismus I
Pathomechanismus II
Physiologische Funktion von Tau-Protein Pathologische Umverteilung von
abnorm phosphoryliertem tau-Protein
in das Soma und die Dendriten
τ P τ P τ P
τ P τ P
τP
τ Pτ P τ P
τ P τ P τ P
τ P τ P
τP
τ τP P
τ P
τP
Ausbildung von
τ bindet an Neurofilamente τ Pbindet nicht an Neurofilamente Neurofibrillenveränderungen
τP
τ P τ P τ P τ P τ Pτ Pτ Pτ P
τ P τ PP
τττττ τ τ τ τ τ τ
P P P P P
τP
τ τP
P
τ P
τ P τ P τ P
Stop des axonalen Axonaler Transport möglich
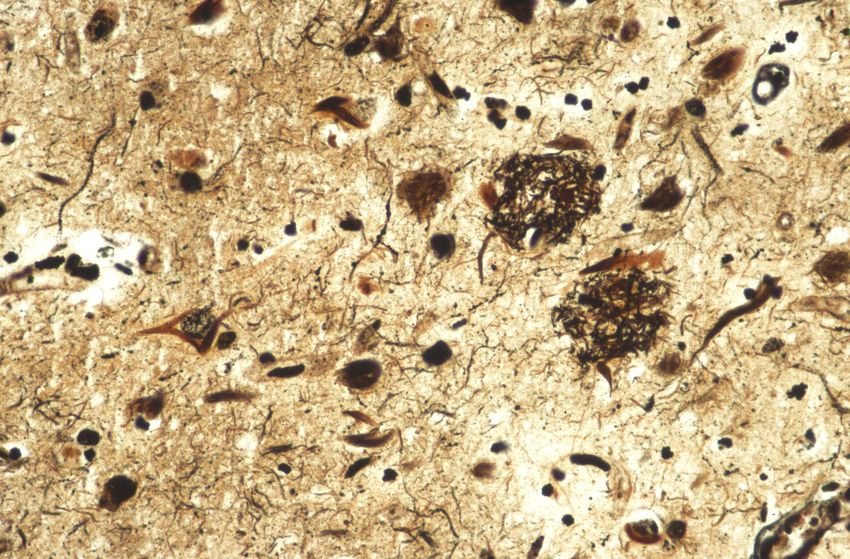
TransportesHistologie und Elektronenmikroskopie
τ P Phosphoryliertes tau:
Tangles, neuritische Plaques,
τ τ P τP τ P
P P P
Neuropilfäden
τ τ P
τ P
τ
τ τP
P
Neuropilfäden Neurofibrillärer
EM: Paired helical filaments
tangle
Neuritischer
Plaque
Gallyas – Färbung (Versilberungstechnik) Immunhistochemie für phosphoryliertes tauUrsachen der Demenz
Alzheimer-Krankheit Vaskuläre
Demenz
Frontotemporale
Demenz
Lewy-Körper-Demenz/
M. Parkinson
Andere
NeurodegenerationenFormen der vaskulären Demenz
- Multi-Infarkt-Demenz (Gesamtvolumen mindestens 50 bis 100 ml)
- „Strategische“ Infarkte: Infarkte in funktionell wichtigen Arealen:
- basales Vorderhirn
- Gyrus angularis
- medialer Thalamus
- Hippokampus
- Mikroangiopathische Demenz:
- Status lacunaris
- subkortikale Leukenzephalopathie (Binswanger-Krankheit)
- granuläre RindenatrophieFrischer Hirninfarkt - Makroskopie − Hirngefäße sind funktionelle Endgefäße − Nach Gefäßverschluss Minderversorgung des Hirngewebes mit Sauerstoff und Glukose
Alter Hirninfarkt - Makroskopie
Hirninfarkt: Mechanismen Verschluss GROSSER Gefäße: Arteriosklerose
Hirninfarkt: Mechanismen
Verschluss kleiner Gefäße: Arteriolosklerose
Risikofaktoren: arterieller Hypertonus, Diabetes mellitus
Status lacunaris
Granuläre RindenatrophieUrsachen der Demenz
Alzheimer-Krankheit Vaskuläre
Demenz
Frontotemporale
Demenz
Lewy-Körper-Demenz/
M. Parkinson
Andere
NeurodegenerationenFronto-temporale Lobärdegeneration (FTLD)
Klinik
Veränderungen der Persönlichkeit, des Sozialverhaltens und der
Sprachproduktion
FTD (Frontotemporale Demenz)
PAX (progressive Apraxie)
SD (Semantische Demenz)
PNFA (Progressive nicht-fluente
Aphasie)
FTD-MND (Frontotemporale
Demenz mit Motoneuron-
Krankheit = ALS)
Epidemiologie
5-15% der Demenzen, beiFronto-temporale Lobärdegeneration (FTLD)
- Erstbeschreiber Arnold Pick = Morbus Pick
- frühe Krankheitsmanifestation im Vergleich
zum M. Alzheimer (40.-50. Lj.)
- familiäre Häufung in 50%
- Klinik: - Interessenlosigkeit
- sexuelle Enthemmung
- Distanzlosigkeit
- eingeschränkte Urteilsfähigkeit
- später Gedächtnisstörungen
Silber-FärbungUrsachen der Demenz
Alzheimer-Krankheit Vaskuläre
Demenz
Frontotemporale
Demenz
Lewy-Körper-Demenz/
M. Parkinson
Andere
NeurodegenerationenMorbus Parkinson I
Definition: Parkinsonismus mit Neuronenverlust (> 50%)
und Lewy-Körpern in pigmentierten Hirnstammkernen
• Prävalenz 1/1000. 1-2% der >65-Jährigen. 2-4% der >85-Jährigen
• zweithäufigste neurodegenerative Krankheit
• mittleres Alter bei Beginn der Krankheit: 61 Jahre
• Dauer der Krankheit: 10-15 Jahre
• in 5% monogenetisch (dominant oder rezessiv)
• in 30% Demenz, in 20% Depression
• zumindest in den ersten Jahren gut mit L-Dopa beeinflussbarMorbus Parkinson II
Rigor
Tremor
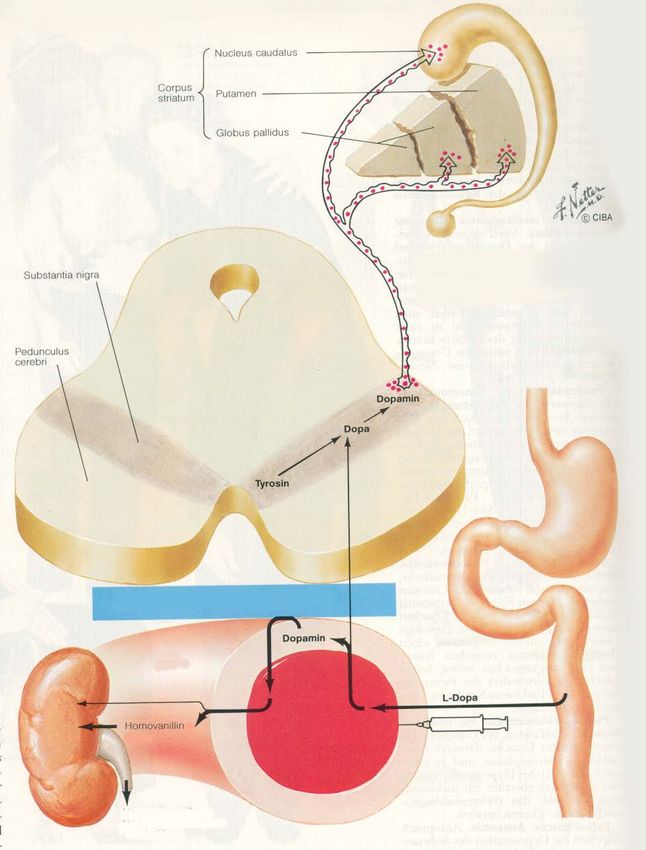
HypokinesePathomechanismus
Pathogenetische
Grundlage:
Reduktion der
dopaminergen
Übertragung von
Substantia nigra zum
StriatumMakroskopie
Kontrolle Parkinson-KrankheitHistologie
Ausfall der pigmentierten Nervenzellen und
Gliose in der Substantia nigraGenetik
Ursachen der Demenz
Alzheimer-Krankheit Vaskuläre
Demenz
Frontotemporale
Demenz
Lewy-Körper-Demenz/
M. Parkinson
Andere
NeurodegenerationenDemenz mit Lewy-Körpern Lewy-Körperchen – eosinophile zytoplasmatische Einschlüsse (Friedrich Lewy: Zur Pathologischen Anatomie der Paralysis agitans. Deutsche Zeitschrift für Nerven- heilkunde 50: 50-55, 1913) Zweithäufigste Ursache einer dementiellen Erkrankung im höheren Lebensalter (>65 Jahre)
Demenz mit Lewy-Körpern - Klinik • Progredienter kognitiver Abbau • Fluktuationen der kognitiven Fähigkeiten • Visuelle Halluzinationen und Parkinson-Symptome
Demenz mit Lewy-Körpern - Histologie
http://de.wikipedia.org/wiki/Lewy-K%C3%B6rper-Demenz
HE-Färbung Immunhistochemie für a-SynucleinUrsachen der Demenz
Alzheimer-Krankheit Vaskuläre
Demenz
Frontotemporale
Demenz
Lewy-Körper-Demenz/
M. Parkinson
Andere
NeurodegenerationenSpongiforme Enzephalopathien (Prion-Krankheiten)
Infektiöse neurodegenerative Krankheiten:
1. Sporadisch-infektiös:
Creutzfeldt-Jakob-Krankheit (90%)
Kuru (ritueller Kannibalismus in Neuguinea)
Scrapie (bei Schafen, Nerzen, Elchen, Maultieren)
Bovine spongiforme Enzephalopathie (BSE)
2. Hereditär (Mutationen im PRNP-Gen):
Creutzfeldt-Jakob-Krankheit (10%)
Gerstmann-Sträussler-Scheinker-Krankheit (v.a. Kleinhirn)
fatale familiäre Insomnie (v.a. Thalamus)Makroskopie
Histologie
Übertragung bei spongiformen Enzephalopathien
• Keine viralen Partikel nachweisbar
• Keine entzündlichen oder immunologischen Veränderungen
• Ablagerungen eines abnormen Proteins, oft in Form von Plaques
Jakob-Creutzfeldt-Krankheit (Neokortex) Gerstmann-Sträussler-Scheinker (Kleinhirn)
• Infektiosität ist mit Plaques assoziiert
• Inaktivierung von Nukleinsäuren reduziert Infektiosität nicht
• Extraktion der Proteinfraktion zerstört InfektiositätDie Prion-Hypothese (Prusiner 1982)
• Das Plaque-Protein ist infektiös
• Das normale (nicht-infektiöse) Prion-Protein (PrPc) wird im Wirts
organismus kodiert und auf Nervenzellen und Lymphozyten exprimiert
• Das infektiöse Prion-Protein (PrPSc) hat die selbe Aminosäuresequenz,
ist jedoch proteinase-resistent und polymerisiert leicht
prn-p Gen prn-p Gen
m-RNA m-RNA
PrPC PrPSc
normales Protein infektiöses Protein
Plaque
normale Zelle infizierte ZelleSpongiforme Enzephalopathien / Creutzfeldt-Jakob-Krankheit:
Neue Variante (vCJD)
Kleinhirn Neocortex
HE
PrP
IHCSpongiforme Enzephalopathien / Creutzfeldt-Jakob-Krankheit:
Neue Variante (vCJD)
Süddeutsche Zeitung Nr. 194 vom 24. August 2001Spekulationen über vCJD während der größten Hysterie 2001 weltweit: 229 vCJD Fälle (Stand: März 2015) kein Fall in Deutschland
Vielen Dank für Ihre Aufmerksamkeit
Sie können auch lesen

















































