EINFLUSS VON ANTI-NMDA-REZEPTOR-NR1- AUTOANTIKÖRPERN BEI APOE4-BEDINGTER CHRONISCHER BEEINTRÄCHTIGUNG DER BLUT-HIRN-SCHRANKE - MPG.PURE
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Aus der Abteilung Klinische Neurowissenschaften
Prof. Dr. med. Dr. med. vet. H. Ehrenreich
des Max-Planck-Instituts für Experimentelle Medizin
in Göttingen
Einfluss von Anti-NMDA-Rezeptor-NR1-
Autoantikörpern bei ApoE4-bedingter
chronischer Beeinträchtigung der
Blut-Hirn-Schranke
INAUGURAL-DISSERTATION
zur Erlangung des Doktorgrades
der Medizinischen Fakultät der
Georg-August-Universität zu Göttingen
vorgelegt von
Maria Regina Zerche
aus
Erfurt
Göttingen 2018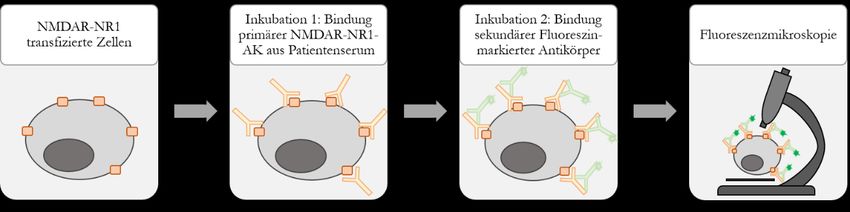
Dekan: Prof. Dr. rer. nat. H.K. Kroemer Referent/in: Prof. Dr. med. Dr. med. vet. H. Ehrenreich Ko-Referent/in: PD Dr. med. J. Liman Drittreferent/in: Prof. Dr. med. T. Meyer Datum der mündlichen Prüfung: 19.07.2018
Hiermit erkläre ich, die Dissertation mit dem Titel "Einfluss von
Anti-NMDA-Rezeptor-NR1-Autoantikörpern bei ApoE4-bedingter
chronischer Beeinträchtigung der Blut-Hirn-Schranke" eigenständig
angefertigt und keine anderen als die von mir angegebenen Quellen
und Hilfsmittel verwendet zu haben.
Göttingen, den …………… ……………………………
(Unterschrift)Teile dieser Dissertation sind bereits in folgenden Fachzeitschriften publiziert: Zerche M*, Weissenborn K*, Ott C, Dere E, Asif AR, Worthmann H, Hassouna I, Rentz- sch K, Tryc AB, Dahm L et al. (2015): Preexisting Serum Autoantibodies Against the NMDAR Subunit NR1 Modulate Evolution of Lesion Size in Acute Ischemic Stroke. Stroke 46, 1180–1186 Hammer C, Zerche M, Schneider A, Begemann M, Nave K-A, Ehrenreich H (2014): Apolipoprotein E4 carrier status plus circulating anti-NMDAR1 autoantibodies: association with schizoaffective disorder. Mol Psychiatry 19, 1054–1056
Inhaltsverzeichnis I
Inhaltsverzeichnis
Abbildungsverzeichnis ..............................................................................................IV
Tabellenverzeichnis .................................................................................................... V
Abkürzungsverzeichnis..............................................................................................VI
1 Einleitung ......................................................................................................... 1
1.1 Blut-Hirn-Schranke ............................................................................................................................. 1
1.1.1 Histologischer Aufbau.......................................................................................................... 1
1.1.2 Ursachen für BHS-Störungen ............................................................................................. 1
1.1.3 Bedeutung der BHS für ZNS-Immunologie .................................................................... 2
1.2 Apolipoprotein E ................................................................................................................................ 3
1.2.1 Genetische Varianz und Isoformen ................................................................................... 3
1.2.2 Physiologische Rolle von ApoE ......................................................................................... 4
1.2.3 Pathophysiologische Rolle der ApoE-Isoformen ............................................................ 5
1.2.4 ApoE4 und BHS-Integrität ................................................................................................. 6
1.3 ZNS-reaktive Autoantikörper ........................................................................................................... 8
1.3.1 Prävalenz in Gesunden/Kranken ....................................................................................... 8
1.3.2 Entstehung von ZNS-reaktiven AK .................................................................................. 8
1.3.3 Potentielle Effekte von ZNS-reaktiven AK ..................................................................... 9
1.4 NMDA-Rezeptor-Autoantikörper.................................................................................................. 10
1.4.1 Rolle des exzitatorischen NMDA-Rezeptors ................................................................. 10
1.4.2 Pathologische Effekte von NMDA-Rezeptor-Autoantikörpern ................................. 11
1.4.3 NMDAR als Target in der Schlaganfall-Therapie .......................................................... 12
1.4.4 Arbeiten zu NMDAR-ABs in der Arbeitsgruppe .......................................................... 12
1.5 Hypothese und Zielsetzung der Arbeit .......................................................................................... 14
1.5.1 ApoE4 und NMDAR-NR1-AK in neuropsychiatrischen Erkrankungen ................. 14
1.5.2 ApoE4 und NMDAR-NR1-AK bei ischämischen Schlaganfällen ............................. 14
2 Material und Methoden .................................................................................. 15
2.1 Material................................................................................................................................................ 15
2.1.1 Geräte .................................................................................................................................... 15
2.1.2 Software ................................................................................................................................ 15
2.1.3 Chemikalien .......................................................................................................................... 16
2.1.4 Verbrauchsmaterialien ........................................................................................................ 16
2.1.5 Enzyme ................................................................................................................................. 17
2.1.6 Kits ........................................................................................................................................ 17
2.1.7 Puffer und Lösungen .......................................................................................................... 17
2.1.8 Primer und DNA-Standards.............................................................................................. 18
2.1.9 Antikörper ............................................................................................................................ 18
2.2 Patientenkollektiv und klinische Datenerhebung ......................................................................... 20Inhaltsverzeichnis II
2.2.1 GRAS-Datenbank ............................................................................................................... 20
2.2.2 Schlaganfall-Projekt ............................................................................................................ 21
2.3 ApoE-Genotypisierung .................................................................................................................... 22
2.3.1 KASP-Genotyping-Assay .................................................................................................. 22
2.3.2 PCR ....................................................................................................................................... 25
2.3.3 Gelelektrophorese ............................................................................................................... 26
2.3.4 Sequenzierung ...................................................................................................................... 26
2.4 Bestimmung des NMDAR-NR1-AK-Status................................................................................. 27
2.4.1 Immunfluoreszenztest ........................................................................................................ 27
2.4.2 Mikroskopische Auswertung ............................................................................................. 28
2.5 Statistische Analyse ........................................................................................................................... 29
3 Ergebnisse....................................................................................................... 31
3.1 Projekt 1: ApoE4 und NMDAR-NR1-AK in neuropsychiatrischen Erkrankungen ............. 31
3.1.1 APOE-Allelfrequenzen und Genotyp-Verteilung im Patientenkollektiv................... 31
3.1.2 Kombination von ApoE4 und NMDAR-NR1-AK im Patientenkollektiv ............... 32
3.1.3 Assoziation zwischen Diagnose und Kombination von ApoE4-Träger-Status
und NMDAR-NR1-AK-Seropositivität .......................................................................... 33
3.1.4 Basalcharakteristika bei Kombination von ApoE4 und NMDAR-NR1-AK............ 34
3.2 Projekt 2: ApoE4 und NMDAR-NR1-AK bei ischämischen Schlaganfällen ......................... 37
3.2.1 Prävalenz von NMDAR-NR1-AK an Tag 1 nach ACM-Infarkt ................................ 37
3.2.1.1 Gesamtprävalenz ....................................................................................................... 37
3.2.1.2 Geschlechtsverteilung seropositiver Patienten ..................................................... 38
3.2.1.3 Titerverteilung an Tag 1 nach ACM-Infarkt ......................................................... 38
3.2.1.4 Altersverteilung der Seroprävalenz......................................................................... 39
3.2.2 Kombination von ApoE4 und NMDAR-NR1-AK im Patientenkollektiv ............... 40
3.2.3 Patientencharakteristika in Abhängigkeit von ApoE4 und NMDAR-NR1-AK ...... 41
3.2.4 Klinischer Verlauf in Abhängigkeit von ApoE4 und NMDAR-NR1-AK ................ 43
3.2.4.1 Outcome-Parameter abhängig nur von NMDAR-NR1-AK-Status ................. 43
3.2.4.2 Evolution der Läsionsgröße anhand von Delta DWI und Delta FLAIR ........ 44
3.2.4.3 Delta DWI und Delta FLAIR nach Immunglobulinklasse ................................ 45
3.2.4.4 NIHSS Score an Tag 7 ............................................................................................. 46
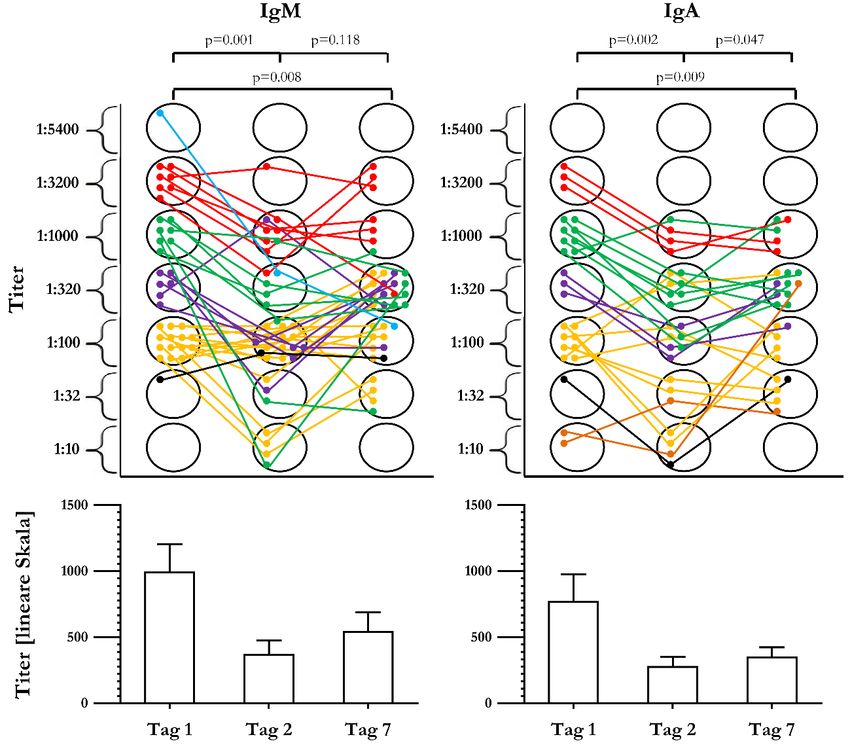
3.2.5 Verlauf der NMDAR-NR1-AK-Titer nach ACM-Infarkt ........................................... 46
4 Diskussion....................................................................................................... 48
4.1 Altersabhängigkeit der NMDAR-NR1-AK-Prävalenz................................................................ 48
4.2 Assoziation zwischen NMDAR-NR1-AK/ApoE4 und schizoaffektiver Störung ................ 49
4.3 Einfluss auf phänotypische Basalcharakteristika .......................................................................... 50
4.4 Einfluss auf den Verlauf nach ACM-Infarkt ................................................................................ 51
4.4.1 Einfluss auf Delta MRT DWI und Delta MRT FLAIR ............................................... 52
4.4.2 Dissoziation zwischen bildgebendem und klinischem Outcome ................................ 53
4.4.3 Unterschiede zwischen den Immunglobulinklassen ...................................................... 54
4.5 NMDAR-NR1-AK-Titerverlauf nach ACM-Infarkt................................................................... 55
4.6 Limitationen von ApoE4 als Indikator der BHS-Integrität ....................................................... 57
4.7 Schlussfolgerung und Ausblick ....................................................................................................... 57Inhaltsverzeichnis III 5 Zusammenfassung .......................................................................................... 59 6 Literaturverzeichnis ........................................................................................ 60 Danksagung ............................................................................................................... 70 Lebenslauf ................................................................... Fehler! Textmarke nicht definiert.
Abbildungsverzeichnis IV
Abbildungsverzeichnis
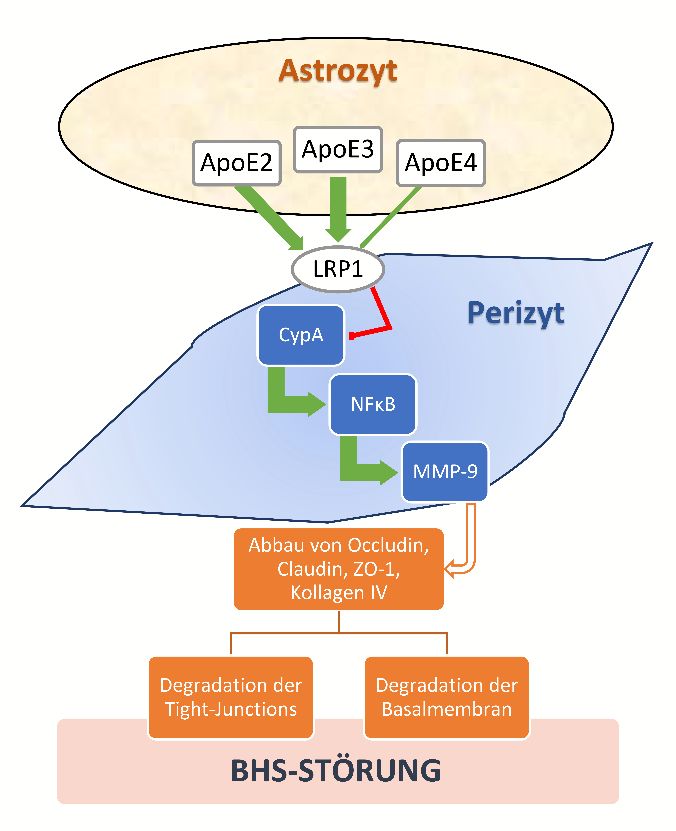
Abbildung 1: ApoE-regulierte LPR1-vermittelte CypA-NFκB-MMP9-Signalkaskade ....................... 7
Abbildung 2: Funktionsprinzip des KASP-Genotyping-Assay ............................................................. 23
Abbildung 3: Grafische Darstellung der Fluoreszenzsignale und SNP-Zuordnung .......................... 25
Abbildung 4: Prinzip des indirekten Immunfluoreszenztests zur NMDAR-NR1-AK
Bestimmung ........................................................................................................................... 28
Abbildung 5: Verteilung der Patienten mit einer Kombination von ApoE4-Träger-Status und
NMDAR-NR1-AK-Seropositivität im Patientenkollektiv ............................................. 32
Abbildung 6: Stapelbalkendiagramm der NMDAR-NR1-AK-Titerverteilung an Tag 1................... 39
Abbildung 7: Altersabhängigkeit der NMDAR-NR1-AK-Seropositivität ........................................... 40
Abbildung 8: Kombination von ApoE4-Träger-Status und NMDAR-NR1-AK-
Seropositivität ........................................................................................................................ 41
Abbildung 9: Bildgebende Outcome-Parameter Delta DWI und Delta FLAIR in Abhängigkeit
von NMDAR-NR1-Seropositivität und ApoE4-Träger-Status..................................... 44
Abbildung 10: Bildgebende Outcome-Parameter Delta DWI und Delta FLAIR in Abhängigkeit
von NMDAR-NR1-Seropositivität und ApoE4-Träger-Status, Aufteilung nach
Immunglobulinklasse............................................................................................................ 45
Abbildung 11: NIHSS Score an Tag 7 in Abhängigkeit von NMDAR-NR1-
Seropositivität und ApoE4-Träger-Status ......................................................................... 46
Abbildung 12: Verlauf der NMDAR-NR1-AK-Titer für IgM und IgA an Tag 1, Tag 2
und Tag 7 nach ACM-Infarkt ............................................................................................. 47V
Tabellenverzeichnis
Tabelle 1: Genotypische und phänotypische Varianz von ApoE ........................................................... 4
Tabelle 2: KASP-Genotyping-Assay PCR-Programm............................................................................ 24
Tabelle 3: Exzitations- und Emissionswellenlängen der KASP-Fluorophore .................................... 24
Tabelle 4: PCR-Programm für ApoE-Genotypisierung ......................................................................... 26
Tabelle 5: Antikörper-Titerbestimmung entsprechend Fluoreszenzsignal.......................................... 29
Tabelle 6: APOE-Allelfrequenzen im Patientenkollektiv ...................................................................... 31
Tabelle 7: ApoE-Genotyp-Verteilung im Patientenkollektiv ................................................................ 31
Tabelle 8: ApoE4-Träger im Patientenkollektiv ...................................................................................... 32
Tabelle 9: Assoziation von ApoE4-Träger-Status und NMDAR-NR1-AK-Seropositivität ............ 33
Tabelle 10: Assoziation zwischen Diagnose und Interaktion von ApoE4-Träger-Status und
NMDAR-NR1-AK-Seropositivität ......................................................................................... 34
Tabelle 11: Basalcharakteristika der Schizophreniepatienten bei Kombination aus ApoE4-
Träger-Status und NMDAR-NR1-AK-Seropositivität ........................................................ 35
Tabelle 12: Basalcharakteristika der schizoaffektiven Patienten bei Kombination aus ApoE4-
Träger-Status und NMDAR-NR1-AK-Seropositivität ........................................................ 36
Tabelle 13: Basalcharakteristika bipolarer Patienten, unipolar depressiver Patienten und
gesunder Kontrollpersonen bei Kombination aus ApoE4-Träger-Status und
NMDAR-NR1-AK-Seropositivität ......................................................................................... 37
Tabelle 14: Prävalenz von NMDAR-NR1-AK an Tag 1 nach ACM-Infarkt ....................................... 38
Tabelle 15: Geschlechtsverteilung NMDAR-NR1-AK-seropositiver Patienten .................................. 38
Tabelle 16: NMDAR-NR1-AK-Titerverteilung an Tag 1 nach ACM-Infarkt...................................... 39
Tabelle 17: Mittleres Lebensalter bei Seropositivität verschiedener Immunglobulinklassen.............. 40
Tabelle 18: Patientencharakteristika in Abhängigkeit von ApoE4- und NMDAR-NR1-AK-
Status ............................................................................................................................................ 42
Tabelle 19: Outcome-Parameter abhängig nur vom NMDAR-NR1-AK-Status ................................. 43Abkürzungsverzeichnis VI Abkürzungsverzeichnis ACM Arteria cerebri media AK Antikörper ANOVA Analysis of Variance ApoE Apolipoprotein E APOE Apolipoprotein-E-Gen ATP Adenosintriphosphat BCR B-Cell-Receptor BHS Blut-Hirn-Schranke Bp Basenpaare C Cytosin CNI Cambridge Neurological Inventory CPZ Chlorpromazin-Äquivalent CRP C-reaktives Protein CypA Cyclophilin A ddNTP Didesoxynukleosidtriphosphat DMSO Dimethylsulfoxid DNA Desoxyribonukleinsäure dNTP Desoxyribonukleosidtriphosphat DSM-IV Diagnostic and Statistical Manual of Mental Disorders, 4. Edition ELISA Enzyme-linked Immunosorbent Assay FAM Carboxyfluoreszein GAF Global Assesment of Functioning GRAS Göttingen Research Association for Schizophrenia HDL High-Density-Lipoprotein HEK Human Embryonic Kidney HEX Hexachlorofluoreszein HLA Human Leukocyte Antigen IDL Intermediate Density Lipoprotein Ig Immunglobulin IgA Immunglobulin A IgE Immunglobulin E IgG Immunglobulin G IgM Immunglobulin M KASP KBiosciences Competitive Allele-Specific PCR LDL Low Density Lipoprotein LRP1 Low Density Lipoprotein Receptor-related Protein 1 MMP9 Matrixmetallopeptidase 9 MRT Magnetresonanztomographie
Abkürzungsverzeichnis VII
MRT DWI Magnetresonanztomographie Diffusion Weighted Image
MRT FLAIR Magnetresonanztomographie Fluid-Attenuated-Inversion-Recovery-
Sequence
MWT Mehrfach-Wortschatz-Intelligenztest
NF-κB Nuclear Factor kappa B
NIHSS National Institut of Health Stroke Scale
NMDA N-Methyl-D-Aspartat
NMDAR N-Methyl-D-Aspartat-Rezeptor
NR1 NMDA-Rezeptor Untereinheit 1
NR2 NMDA-Rezeptor Untereinheit 2
NVE Neurovaskuläre Einheit
OR Odds ratio
PANSS Positive And Negative Syndrome Scale
PBS Phosphat Buffered Saline
PCR Polymerase Chain Reaction
PKC Proteinkinase C
Q Quencher
ROX Carboxy-X-Rhodamine
SNP Single-Nucleotide-Polymorphism
T Thymin
TAE TRIS-Acetat-EDTA-Puffer
TG Trigyleride
TJ Tight Junction
VLDL Very-Low-Density-Lipoprotein
ZO-1 Zonula Occludens-1 Protein
ZNS Zentrales Nervensystem1 Einleitung 1 1 Einleitung 1.1 Blut-Hirn-Schranke Die Blut-Hirn-Schranke (BHS) stellt eine physiologische Barriere zwischen Blut und Extra- zellularraum des zentralen Nervensystems (ZNS) dar und garantiert durch ihren Aufbau mittels verschiedener Barrieremechanismen die Unzugänglichkeit des ZNS gegenüber höhermolekularen hydrophilen Stoffen (Abbott et al. 2006). Dies schützt das ZNS vor toxischen Substanzen und ermöglicht so die Aufrechterhaltung einer stabilen Umgebung für optimale neuronale Funktion und synaptische Übertragung (Abbott et al. 2010). 1.1.1 Histologischer Aufbau Histologisch weist der Aufbau der zerebralen Gefäßstrukturen wesentliche Unterschiede zu peripheren Kapillaren auf, welche für die Integrität der BHS zwingend erforderlich sind. Neben Endothelzellen sind Perizyten und Astrozyten sowie Elemente der Extrazellular- matrix und Neurone am Aufbau der BHS beteiligt. Der funktionelle Zusammenschluss dieser Strukturen wird als neurovaskuläre Einheit bezeichnet (Sandoval und Witt 2008). Die Endothelzellen und ihre Basallamina stellen die erste Barriere der BHS dar und unterscheiden sich von peripheren Endothelien durch fehlende Fenestrierung sowie die Ausbildung von Tight Junctions (TJ), welche als apikale transmembrane Proteinkomplexe eine feste Verbindung zwischen den Endothelzellen schaffen und parazelluläre Permeabili- tät limitieren (Hawkins und Davis 2005; Abbott et al. 2010). Die die Endothelzellen bedeckenden Perizyten und Astrozyten tragen ebenfalls zur Stabilität der Gefäße bei und regulieren durch Interaktion mit dem Endothel mikrovaskuläre Permeabilität, zerebralen Blutfluss und Wasserhomöostase sowie Formation und Aufrechterhaltung der BHS (Von Tell et al. 2006; Abbott et al. 2006). Die aus Strukturproteinen aufgebaute Extra- zellularmatrix dient als Verankerung der Zellen über Adhäsionsmoleküle und reguliert interzelluläre Kommunikation (Sandoval und Witt 2008). 1.1.2 Ursachen für BHS-Störungen BHS-Störungen können je nach Ausmaß und Dauer in lokale oder globale sowie akute oder chronische Barrierestörungen eingeteilt werden (Rosenberg 2012). Prinzipiell kann dabei eine Fehlfunktion jedes Bestandteils der neurovaskulären Einheit ursächlich für die Schrankenstörung sein (Sandoval und Witt 2008). Ätiologisch kommen, wie im Review von Abbott et al. 2010 beschrieben, zahlreiche ZNS- Pathologien in Betracht: Hierzu gehören unter anderem Ischämien und Hypoxien im Rahmen von Schlaganfällen, infektiöse und entzündliche Prozesse, Schädel-Hirn-Traumata,
1 Einleitung 2 epileptische Anfälle, Geburtskomplikationen, Autoimmunerkrankungen und paraneoplasti- sche Vorgänge (Abbott et al. 2010; Brimberg et al. 2015). Des Weiteren besteht eine Asso- ziation zwischen einer BHS-Dysfunktion mit neurologischen Erkrankungen wie dem Morbus Alzheimer (Zlokovic 2011), dem idiopathischen Parkinson-Syndrom (Desai et al. 2007) oder der Multiplen Sklerose (Ortiz et al. 2014), wobei jedoch nicht abschließend geklärt ist, inwiefern die Permeabilitätserhöhung der BHS Ursache oder Folge dieser Krankheitsbilder darstellt (Stanimirovic und Friedman 2012). Chronischer Nikotin- oder Drogenkonsum - insbesondere von Metamphetamin oder Kokain - sowie inhalative Anästhetika und metabolische Erkrankungen wie Diabetes mellitus Typ II werden eben- falls als Risikofaktoren diskutiert (Sajja et al. 2016; Acharya et al. 2015; Yoo et al. 2016). Im Rahmen dieser Arbeit ist jedoch im Besonderen auf zwei Faktoren mit Einfluss auf die BHS-Integrität einzugehen: Apolipoprotein E (ApoE) stellt einen genetischen Faktor für eine chronische Barrierestörung dar, da die Expression seiner Isoform ApoE4 durch Verminderung der TJ-Assembly mit einer chronischen Semipermeabiliät der BHS assoziiert ist (Bell et al. 2012). Der Mechanismus ist im Kapitel 1.2.4 genauer erläutert. Ischämische Schlaganfälle initiieren hingegen eine sofortige transiente BHS-Dysfunktion (Brouns und Deyn 2009). Durch die plötzliche Minderperfusion kommt es im betroffenen Areal zu akutem ATP- und Sauerstoffmangel und damit zu einer anaeroben Stoffwechsel- lage mit Laktatakkumulation und Bildung freier Sauerstoffradikale (Sandoval und Witt 2008). Dies hat eine Störung der Homöostase und des Ionengleichgewichts mit Zusammenbruch des Membranpotentials, endothelialer Schwellung und der Induktion inflammatorischer Prozesse zur Folge (Sandoval und Witt 2008; Krueger et al. 2015). Die Aktivierung verschiedener Signalkaskaden durch reaktive Sauerstoffspezies mit nach- folgender Veränderung der Expression der TJ-Proteine Claudin-5 und Occludin führt zu einer Disruption der TJ mit Erhöhung des parazellulären Flusses (Schreibelt et al. 2007). 1.1.3 Bedeutung der BHS für ZNS-Immunologie Die komplexen Barrieremechanismen der BHS vermitteln durch eine Limitierung des Zugangs von Zellen und Mediatoren des Immunsystems eine immunologische Sonder- stellung des zentralen Nervensystems (Muldoon et al. 2013). Obwohl sich in den vergangenen Jahren zeigte, dass die Interaktion zwischen Immun- und Nervensystem auch unter physiologischen Bedingungen eine deutlich wichtigere Rolle spielt als zunächst angenommen, ist es zirkulierenden Leukozyten bei intakter BHS nur in Ausnahmefällen möglich, das ZNS-Parenchym zu erreichen und dort eine Immunantwort hervorzurufen (Marin und Kipnis 2017; Muldoon et al. 2013; Szczepanik 2011). Diese mangelnde Interaktion führt zu einer fehlenden negativen Selektion und damit zur Persistenz von autoreaktiven B-Zellen, die ZNS-reaktive Antikörper (AK) produzieren. Wichtiger Teil der im Säuglingsalter stattfindenden Entwicklung der adaptiven Immunant- wort ist die periphere Toleranzentwicklung, welche die Unterscheidung des Immunsystems
1 Einleitung 3 zwischen selbst und fremd ermöglicht (Pelanda und Torres 2012). Aufgrund der BHS- bedingten Isolation des ZNS kann diese Selbsttoleranz gegenüber ZNS-Antigenen nicht entstehen (Diamond et al. 2013). Die BHS erfüllt damit zwei wichtige Funktionen. Zum einen schirmt sie ZNS-Antigene von der peripheren Zirkulation ab und limitiert so ihre Teilnahme an der negativen Selektion unreifer B-Zellen. Zum anderen schützt sie das Gehirn vor dadurch entstehenden ZNS-reaktiven AK (Brimberg et al. 2015). Genauer wird auf diesen Prozess im Rahmen der Entstehung und potentiellen Effekte von ZNS- reaktiven AK in Kapitel 1.3.2 und 1.3.3 eingegangen. Die ZNS-Immunantwort wird daher insbesondere durch Mechanismen des angeborenen Immunsystems vermittelt, wobei Mikroglia-Zellen, die als ZNS-Makrophagen fungieren, eine bedeutende Rolle zukommt (Kettenmann et al. 2011; Yamasaki et al. 2014; Marin und Kipnis 2017). Im Vergleich zu peripheren Makrophagen zeigen sie jedoch eine deutlich geringere Kapazität hinsichtlich Antigenpräsentation und Phagozytose, was ebenfalls zu einer abgeschwächten zentralnervösen Immunantwort und Toleranzentwicklung beiträgt (Prinz et al. 2011; Marin und Kipnis 2017). 1.2 Apolipoprotein E Apolipoprotein E ist ein aus 299 Aminosäuren bestehendes, 34 kDa schweres Glyko- protein, welches durch das auf dem langen Arm des Chromosom 19 lokalisierte und aus vier Exons und drei Introns bestehende APOE-Gen codiert wird (Das et al. 1985). 1.2.1 Genetische Varianz und Isoformen Das humane APOE-Gen weist einen genetischen Polymorphismus auf, wodurch es zur Expression verschiedener Isoformen des Apolipoproteins E kommt. Dabei werden drei Isoformen unterschieden, welche sich jeweils in einer Aminosäure unterscheiden und entsprechend ihrer Ladungsunterschiede und Positionierung nach isoelektrischer Fokussie- rung als ApoE2, ApoE3 und ApoE4 bezeichnet werden (Utermann et al. 1977). Ursache dieser Varianz ist das Auftreten drei verschiedener Allele am Genlokus des APOE durch die beiden Einzelnukleotid-Polymorphismen (engl. Single-Nucleotide- Polymorphism, SNP) rs429358 und rs7412 in Exon 4 des Gens (Koch et al. 2002; Seripa et al. 2011). An beiden Positionen findet sich je nach Genotyp entweder Cytosin (C) oder Thymin (T), was im Rahmen der Translation zu einer Aminosäure-Substitution zwischen Cystein und Arginin an Position 112 und 158 des Proteins führt. Es entstehen die drei Allele ε2, ε3 und ε4 mit den entsprechenden Isoformen ApoE2, ApoE3 und ApoE4 (Yu et al. 2007; Seripa et al. 2011). Trotz deutlicher Varianz der Allelfrequenzen in verschiedenen Populationen stellt ε3 mit einer Allelfrequenz von rund 77 % den häufigsten Haplotyp dar, gefolgt von ε4 mit 15 % und ε2 mit 8 % (Mahley und Rall, JR 2000). Zusammenfassend ist dies in Tabelle 1 dargestellt.
1 Einleitung 4 Tabelle 1: Genotypische und phänotypische Varianz von ApoE Zusammenfassende Darstellung der genotypischen und phänotypischen Varianz von ApoE aufgrund des Polymorphismus am Genlokus des APOE. Die angegebenen Allelfrequenzen entsprechen den beobachteten Frequenzen in nordamerikanischen Populationen europäischer Abstammung. Weltweit wurden Schwan- kungen von 1-15 % für ε2, 50-90 % für ε3 und 5-35 % für ε4 beobachtet (Mahley und Rall, JR 2000). APOE-Gen Apolipoprotein E SNP SNP Allelname Allelfrequenz Aminosäure Aminosäure Isoform rs429358 rs7412 Position 112 Position 158 T T ε2 ~8 % Cystein Cystein ApoE2 T C ε3 ~77 % Cystein Arginin ApoE3 C C ε4 ~15 % Arginin Arginin ApoE4 Es ergeben sich somit sechs mögliche genotypische und phänotypische Kombinationen, von denen je drei homozygot (ApoE2/E2, ApoE3/E3, ApoE4/E4) und drei heterozygot (ApoE2/E3, ApoE2/E4, ApoE3/E4) sind (Mahley 1988). Neben den drei Hauptisoformen sind über 30 weitere ApoE-Varianten bekannt, welche jedoch mit deutlich niedrigeren Allelfrequenzen auftreten und daher nur eine unter- geordnete Rolle spielen (Knijff et al. 1994). 1.2.2 Physiologische Rolle von ApoE ApoE ist ein von zahlreichen Geweben synthetisiertes multifunktionales Protein mit Bedeutung für diverse physiologische Prozesse. Zum Großteil von Hepatozyten expri- miert, nimmt es als Bestandteil von Chylomikronen, Very-Low-Density-Lipoproteinen (VLDL) und High-Density-Lipoproteinen (HDL) insbesondere im Fettstoffwechsel des Menschen eine bedeutende Rolle ein (Mahley 1988). Als hochaffiner Ligand für LDL- und LRP1-Rezeptoren vermittelt es die Endozytose von Plasma-Lipoproteinen und wirkt somit als Regulator der Lipoprotein-Konzentration, was die Assoziation zwischen ApoE- Dysfunktionen und Hyperlipoproteinämie erklärt (Getz und Reardon 2009). Des Weiteren konnte ein protektiver Effekt auf atherosklerotische Prozesse gezeigt werden, da ApoE den Cholesterintransport aus lipidgefüllten Makrophagen der arteriellen Tunica Media fördert (Yancey et al. 2007). Neben der Leber, wo 75 % der Gesamtsynthese des Körpers erfolgt (Mahley 2016), wird ApoE vor allem im Gehirn exprimiert, wobei die Produktion hauptsächlich in Astrozyten (Boyles et al. 1985), jedoch auch in Oligodendrozyten, Mikroglia und Neuronen erfolgt (Xu et al. 2006). Die lokale ZNS-eigene Produktion ist insbesondere aufgrund der Undurchläs- sigkeit der BHS gegenüber systemischen Apolipoproteinen von Bedeutung (Mahley 2016). ApoE dient im ZNS als Cholesterin-Transport-Protein zwischen Astrozyten und Neuronen und spielt eine wichtige Rolle im Rahmen cholesterinabhängiger Prozesse wie
1 Einleitung 5 neuronaler Regeneration, Neuritenwachstum und Synaptogenese, da es die Umverteilung von Cholesterin und Phospholipiden ermöglicht (Hauser et al. 2011; Zlokovic 2013). Zudem ist ApoE für die Aufrechterhaltung der BHS-Integrität von Bedeutung (siehe Kapitel 1.2.4) (Bell et al. 2012). ApoE wird von zahlreichen weiteren Zellen exprimiert, zu denen Adipozyten, Macro- phagen und glatte Muskelzellen gehören (Huang et al. 2006; Xu et al. 2006). Neben seiner Rolle im Fettstoffwechsel sind verschiedene lipidunabhängige Funktionen des Proteins bekannt, zu denen die Inhibition der Proliferation glatter Muskelzellen, anti- inflammatorische Effekte auf T-Zellen und verminderte Zytokinproduktion, sowie eine antioxidative Wirkung gehören (Hui und Basford 2005; Tenger und Zhou 2003; Getz und Reardon 2009). 1.2.3 Pathophysiologische Rolle der ApoE-Isoformen Die drei ApoE-Isoformen unterscheiden sich nur in jeweils einer Aminosäure, was jedoch durch eine Veränderung struktureller und funktioneller Eigenschaften zu einer patho- physiologischen Rolle im Rahmen verschiedener Krankheitsbilder führt (Mahley 2016). Die mit einer Allelfrequenz von 77 % häufigste Isoform ApoE3 stellt die voll funktionale Normvariante dar (Mahley und Rall, JR 2000). ApoE2 weist hingegen, bedingt durch eine Seitenketten-Konformationsänderung im Bereich der Rezeptorbindungsdomäne, eine deutlich niedrigere Bindungsaffinität gegenüber dem LDL-Rezeptor auf, was zu einer verminderten Clearance von VLDL, Intermediate-Density-Lipoproteinen (IDL) und Chylomikronen Remnants und damit zu einer Dysregulation der Cholesterin-Homöostase führt (Wilson et al. 1991; Wilson et al. 1994). Es resultiert eine Störung des Fettstoffwech- sels mit einem deutlich erhöhten Risiko der Entwicklung einer Typ-III-Hyperlipo- proteinämie und Atherosklerose, insbesondere bei homozygoten ApoE2-Trägern (Mahley et al. 2009). ApoE4 ist - aufgrund einer bevorzugten Bindung an Triglycerid-reiche VLDLs und einer konsekutiven Herunterregulation von LDL-Rezeptoren - ebenfalls mit erhöhten LDL- Konzentrationen und einem erhöhten Risiko für Atherosklerose assoziiert (Weintraub et al. 1987; Li et al. 2013). Im Vordergrund steht bei dieser Isoform jedoch vor allem seine Rolle als Risikofaktor der late-onset-Alzheimer-Erkrankung, wobei ApoE4 sowohl mit einem jüngeren Erkrankungsalter als auch einer rascheren Progression und auch Schwere der Erkrankung assoziiert ist (Corder et al. 1993; Hultman et al. 2013). Die Ursache der beschleunigten neuronalen Degeneration bei ApoE4-Trägern liegt vermutlich unter anderem in einer erhöhten neuronalen ApoE-Produktion in Stresssituationen (Xu et al. 2006). Geschädigte Neurone synthetisieren verstärkt ApoE, um eine Lipidumverteilung zur Unterstützung von Reparaturmechanismen zu ermöglichen (Zlokovic 2013). Das abnorm konformierte ApoE4 wird jedoch proteolytischen Abbauprozessen unterzogen, wodurch neurotoxische Fragmente entstehen, die im Zytosol zu mitochondrialer Dysfunktion,
1 Einleitung 6 tau-Phosphorylierung, Bildung neurofibrillärer Bündel und letztlich dem Zelltod führen (Chen et al. 2011; Huang et al. 2001; Mahley 2016). Transgene ApoE4 überexprimierende Mäuse weisen einen Untergang hippocampaler Neurone, synaptische Defizite und vermin- dertes Neuritenwachstum auf, was mit einer Beeinträchtigung von Kognition, Lernen und Gedächtnis einhergeht (Hauser et al. 2011). Daneben ist ApoE4 mit einem schlechteren klinischen Outcome nach Schädel-Hirn- Traumata und Schlaganfällen assoziiert sowie mit neurodegenerativen Erkrankungen wie Frontotemporaldemenz und Multipler Sklerose (Friedman et al. 1999; McCarron et al. 1999; Agosta et al. 2009; Chapman et al. 2001). 1.2.4 ApoE4 und BHS-Integrität Einen weiteren Erklärungsansatz für die pathophysiologische Relevanz von ApoE4 im Rahmen zerebrovaskulärer Erkrankungen stellt die Rolle des Proteins für die Aufrecht- erhaltung der BHS dar. Als hoch affiner Ligand des LRP1-Rezeptors reguliert ApoE verschiedene intrazelluläre Signalkaskaden mit Einfluss auf die Integrität von TJ, Basalmembran und Perizyten (Halliday et al. 2016). Dabei ist insbesondere der von Bell et al. 2012 beschriebene proinflammatorische CypA-NFκB-MMP9-Signalweg von Bedeutung, welcher in transgenen ApoE-Knockout- und ApoE4-Mäusen, nicht aber ApoE2 oder ApoE3 exprimierenden Tieren, zu einer altersabhängigen progressiven BHS-Störung führt. Abbildung 1 zeigt eine schematische Darstellung des Einflusses der ApoE-Isoformen auf diesen Signalweg. Im physiologischen Fall inhibiert das von Astrozyten synthetisierte ApoE LRP1-vermittelt die Akkumulation von Cyclophilin A (CypA) in Perizyten. Aufgrund der deutlich geringeren LRP1-Rezeptor-Affinität von ApoE4 im Vergleich zu den beiden anderen Isoformen ist bei ApoE4-Trägern diese Inhibition jedoch vermindert (Bell et al. 2012; Halliday et al. 2013; Halliday et al. 2016). Es resultiert eine CypA-induzierte Aktivierung des Transkriptionsfaktors Nuclear Factor kappa B (NF-κB), was zu einer verstärkten Expression der proteolytischen Matrix-Metalloproteinase-9 (MMP-9) führt. Dies wiederum zieht den beschleunigten Abbau der Substrate von MMP-9 nach sich, zu denen die TJ-Proteine Occludin, Claudin und Zonula occludens-1 Protein (ZO-1) sowie das für den Aufbau der Basalmembran wichtige Strukturprotein Kollagen IV gehören (Bell et al. 2012). Ergebnis ist die Degradation von TJs und Basalmembran und damit eine BHS-Störung mit Erhöhung des parazellulären Flusses und Erniedrigung des transendo- thelialen elektrischen Widerstandes (Bell et al. 2012; Nishitsuji et al. 2011). Dies konnte wiederholt in Tierversuchen durch eine Erhöhung der Durchlässigkeit der BHS gegenüber injizierten Farbstoffen gezeigt werden (Fullerton et al. 2001; Hafezi-Moghadam et al. 2007; Methia et al. 2001).
1 Einleitung 7
ApoE2 ApoE3 ApoE4
LRP1
CypA
NFκB
MMP-9
Abbau von Occludin,
Claudin, ZO-1,
Kollagen IV
Degradation der Degradation der
Tight-Junctions Basalmembran
Abbildung 1: ApoE-regulierte LPR1-vermittelte CypA-NFκB-MMP9-Signalkaskade
Dargestellt ist der Einfluss der verschiedenen ApoE-Isoformen auf die BHS-Integrität über die Low-Density-
Lipoprotein-Rezeptor-related-Protein-1-(LRP1)-vermittelte Hemmung der proinflammatorischen CypA-
NFκB-MMP9-Signalkaskade (Bell et al. 2012). In Astrozyten exprimiertes ApoE2 oder ApoE3 aktiviert auf
Perizyten lokalisierte LRP1-Rezeptoren, was durch die resultierende Inhibition von Cyclophilin A (CypA) zu
einer geringeren Expression von Nuclear Factor kappa B (NFκB) und Matrix-Metalloproteinase 9 (MMP9)
führt. Aufgrund geringerer LRP1-Rezeptoraffinität von ApoE4 ist diese Regulation bei ApoE4-Expression
herabgesetzt, so dass es zum MMP-9 vermittelten Abbau von Occludin, Claudin, ZO-1 und Kollagen IV und
damit zur Degradation von Tight-Junctions und der Basalmembran kommt. Es resultiert eine BHS-Störung.
Abbildung modifiziert nach Nigro et al. 2013, Verwendung nach Creative Commons CC BY 3.0 Lizenz.
Über einen weiteren, ebenfalls LRP1-vermittelten Signalweg ist ApoE darüber hinaus für
die Ausbildung der TJ von Bedeutung. Durch Bindung an den LRP1-Rezeptor erfolgt eine
Aktivierung der Proteinkinase C (PKC), welche das TJ-Protein Occludin phosphoryliert.
Diese Phosphorylierung ist für den Aufbau der TJ unerlässlich, bei ApoE4-Trägern jedoch
aufgrund der geringeren LRP1-Bindungsaffinität wiederum vermindert, was zu einer
höheren BHS-Permeabilität von ApoE4-Trägern führt (Nishitsuji et al. 2011).
ApoE ist somit integraler Bestandteil der Aufrechterhaltung der BHS, weshalb bei ApoE4-
Trägern von einer chronischen Beeinträchtigung der Barrierefunktion mit resultierender
Semipermeabilität der BHS auszugehen ist (Bell et al. 2012; Halliday et al. 2013).1 Einleitung 8 1.3 ZNS-reaktive Autoantikörper ZNS-reaktive Autoantikörper sind in B-Zellen produzierte, zirkulierende Autoantikörper gegen Antigene, die nur im ZNS und nicht in anderen Geweben auftauchen. Aufgrund der erläuterten Barrieremechanismen zwischen Blut und ZNS-Parenchym spielt die Integrität der BHS sowohl im Rahmen ihrer Entstehung als auch ihrer Wirkung eine bedeutende Rolle (Diamond et al. 2009). 1.3.1 Prävalenz in Gesunden/Kranken In den vergangenen Jahren konnte eine große Diversität dieser AK gegen verschiedene Antigene identifiziert werden (Levin et al. 2010), denen zunächst vornehmlich aufgrund ihres vermehrten Auftretens im Zusammenhang mit ZNS-Pathologien verstärkte Aufmerksamkeit zukam. Beispiele hierfür sind der neuropsychiatrische Lupus erythematodes oder die NMDA-Rezeptor-Antikörper-Enzephalitis (Diamond et al. 2009; Dalmau et al. 2008; Diamond et al. 2013). Es zeigte sich jedoch, dass sich die Prävalenz ZNS-reaktiver AK nicht auf erkrankte Personen beschränkt, sondern diese auch im Serum von gesunden Probanden in vergleichbarem Maße vorhanden sind (Levin et al. 2010; Coutinho et al. 2014). So findet sich sowohl in gesunden als auch neuropsychiatrisch erkrankten Personen eine Seroprävalenz von altersabhängig bis zu 20 % von AK gegen die NMDA-Rezeptor- Untereinheit NR1 (Hammer et al. 2014a; Dahm et al. 2014). Dies konnte wiederholt repliziert werden (Steiner et al. 2013; Steiner et al. 2014; Busse et al. 2014). 1.3.2 Entstehung von ZNS-reaktiven AK Zur Erklärung dieser hohen Seroprävalenz ZNS-reaktiver AK trotz der erläuterten Barrieremechanismen der BHS ist eine genauere Betrachtung der Entstehungsmechanis- men ZNS-reaktiver AK erforderlich. Die Bildung ZNS-reaktiver AK findet vor allem im Rahmen von Autoimmun- erkrankungen, als Kreuzreaktion nach Exposition mit exogenen Antigenen und im Zusammenhang mit paraneoplastischen Syndromen statt (Diamond et al. 2013). Erfolgt sie, wie beispielsweise beim neuropsychiatrischen Lupus Erythematodes, im Rahmen einer Autoimmunerkrankung, ist eine Assoziation mit spezifischen Human Leucocyte Antigen (HLA) Haplotypen und weiteren Risikoallelen zu erwarten (Lee et al. 2015). Doch auch ohne genetische Suszepitibilität ist durch Kreuzreaktionen die Initiation einer autoreaktiven Immunantwort gegenüber ZNS-Strukturen möglich. Exposition gegenüber exogenen Mikroben- und Nahrungsmittelantigenen kann, beispielsweise im Rahmen von Erkrankungen wie der Chorea minor Sydenham, über molekulare Mimikry ebenfalls zur Ausbildung autoreaktiver Antikörper führen (Kirvan et al. 2006). Bei dieser Spätmanifestation des durch Streptokokken ausgelösten Rheumatischen Fiebers, kommt es durch Kreuzreaktivität zwischen Antikörpern gegen Streptokokken und striatalen
1 Einleitung 9 Neuronen zur Ausbildung von Anti-Basalganglien-Antikörpern (Kirvan et al. 2006). Des Weiteren sind zahlreiche antikörpervermittelte paraneoplastische Syndrome des ZNS beschrieben, welche unter anderem bei Teratomen, kleinzelligen Lungenkarzinomen, sowie Mamma- und Ovarialkarzinomen auftreten (Dalmau et al. 2007; Bataller et al. 2004; Rojas- Marcos et al. 2003). Gegen Tumorantigene gerichteten Antikörper zeigen sich dabei kreuz- reaktiv gegenüber strukturell homologen ZNS-Antigenen (Diamond et al. 2013). Die hohe Prävalenz ZNS-reaktiver AK in gesunden Probanden ist jedoch nicht durch ihr Auftreten im Rahmen von Pathologien zu erklären. Derzeit wird vor allem ihre Bildung in autoreaktiven B-Zellen diskutiert, welche durch fehlende negative Selektion im ZNS persistieren (Diamond et al. 2009). Bei der peripheren Toleranzentwicklung des Immunsystems werden T- und B-Zellen, die gegen körpereigene Antigene gerichtet sind, normalerweise eliminiert. Hierfür ist die Antigen-vermittelte Triggerung des B-Zell- Rezeptors (BCR) erforderlich (Janeway et al. 2001). Aufgrund der Barrieremechanismen der BHS, deren Bildung in der Embryonalentwicklung zeitlich vor der Ausbildung des B-Zell-Repertoires erfolgt, ist dies jedoch bei autoreaktiven B-Zellen gegen ZNS- Strukturen nicht möglich (Saunders et al. 1999; Daneman et al. 2010). Vor allem Neurone und Astrozyten exprimieren zahlreiche Antigene, die in anderen Geweben des Körpers nicht oder nur in anderer Konformation auftreten, so dass B-Zellen, die AK gegen diese Antigene produzieren, während der Toleranzentwicklung nicht untergehen, sondern zu immunkompetenten Zellen heranreifen (Diamond et al. 2013; Brimberg et al. 2015). Dies könnte die wiederholt gefundene hohe Prävalenz ZNS-reaktiver AK in gesunden Proban- den erklären und legt sogar die Hypothese nahe, dass alle Individuen ZNS-reaktive AK entwickeln (Levin et al. 2010). 1.3.3 Potentielle Effekte von ZNS-reaktiven AK Nicht nur die Entstehung, sondern auch potentielle Effekte ZNS-reaktiver AK sind von der Integrität der BHS abhängig, da durch deren Barrieremechanismen die Passage der AK ins ZNS-Parenchym verhindert wird und diese so gegenüber ihren Zielantigenen abgeschirmt sind (Brimberg et al. 2015; Diamond et al. 2009). Ihre potentiell pathogenen Effekte treten daher nur bei einer Beeinträchtigung der BHS auf (Diamond et al. 2013; Hammer et al. 2014a). Zirkulierende AK mit pathogenem Potential vermitteln ihre Wirkung somit in zwei Phasen. Zunächst müssen sie Zugang zum ZNS-Parenchym erhalten. Im zweiten Schritt verursachen sie eine direkte oder Mikroglia-vermittelte Schädigung von Astrozyten und Neuronen (Brimberg et al. 2015; Nestor et al. 2016). Dies erfolgt meist durch Bindung an Membranrezeptoren, was zur Aktivierung intrazellulärer Signalkaskaden, Rezeptor- Internalisierung oder Blockade normaler zellulärer Interaktionen führt (Diamond et al. 2009). Ergebnis ist je nach Konzentration des Antikörpers entweder eine Modulation neuronaler Funktionen oder die Initiation apoptotischer Vorgänge (Diamond et al. 2013).
1 Einleitung 10 Es sind zahlreiche mit ZNS-reaktiven AK assoziierte Pathologien bekannt, die sich als neurologische und neuropsychiatrische Erkrankungen mit Veränderungen von Vigilanz, Verhalten oder Kognition, psychotischen Episoden, epileptischen Anfällen, oder Bewegungsstörungen manifestieren können (Kowal et al. 2004; Abdel-Nasser et al. 2008; Dalmau et al. 2007; Kirvan et al. 2006). Beispiele sind, neben den bereits erwähnten, die Neuromyelitis optica, die limbische Enzephalitis, Hashimoto-Enzephalitis und ZNS- Symptome im Rahmen der glutensensitiven Enteropathie (Jarius et al. 2008; Vincent et al. 2004; Gini et al. 2008; Alaedini et al. 2007). Es gilt daher als bestätigt, dass ZNS-reaktive AK eine signifikante Rolle für verschiedene ZNS-Pathologien spielen (Diamond et al. 2013). Insbesondere aufgrund ihrer hohen Prävalenz ist jedoch auch eine physiologische Funktion der AK zu diskutieren. Klassische Funktion von AK ist ihre Assistenz in der Beseitigung zellulärer Abbauprodukte, sowie die Eradikation und Neutralisation von Pathogenen und Toxinen (Janeway, JR et al. 2001). Vor allem für die letzteren beiden ist eine große Diversität von AK gegen verschiedene Antigene erforderlich. Eine fehlende negative Selektion gegenüber ZNS-Antigenen könnte daher wichtig für den Erhalt dieser Vielfalt sein und den Organismus so durch ein erweitertes B-Zell-Repertoire befähigen, einen effektiven Schutz vor Infektionen und Toxinen zu bieten (Brimberg et al. 2015; Diamond et al. 2013). Weiterhin ist es möglich, dass auch den AK selbst physiologische Funktionen zu kommen und sie, beispielsweise im Rahmen von Abbau- und Reparaturprozessen, für die ZNS-Homöostase von Bedeutung sind. In diesem Fall wäre von einer physiologischen Autoimmunität zu sprechen (Levin et al. 2010). 1.4 NMDA-Rezeptor-Autoantikörper NMDA-Rezeptor-Autoantikörper sind Antikörper, deren Antigen der postsynaptische exzitatorische N-Methyl-D-Aspartat-Rezeptor (NMDAR) ist. Ihre Seroprävalenz ist vor allem im Rahmen paraneoplastischer Syndrome beschrieben und dabei insbesondere mit Teratomen assoziiert (Dalmau et al. 2007). 1.4.1 Rolle des exzitatorischen NMDA-Rezeptors Der ubiquitär im Gehirn exprimierte NMDA-Rezeptor ist als ligandengesteuerter ionotroper Rezeptor des glutamatergen Systems integraler Teil exzitatorischer synaptischer Signalübertragung (Diamond et al. 2009; Li und Tsien 2009). Es handelt sich um einen tetrameren Rezeptor, bestehend aus jeweils zwei NR1 und NR2 Untereinheiten, welcher neben der Glutamat-Bindungsstelle weitere Bindungsstellen für den Coagonisten Glycin, sowie für die Rezeptoraktivität modulierende Faktoren aufweist (Cull-Candy und Leszkiewicz 2004; Kalia et al. 2008).
1 Einleitung 11 Besondere Bedeutung kommt dem NMDA-Rezeptor aufgrund seiner Leitfähigkeits- erhöhung durch wiederholte Reizung und Depolarisation der postsynaptischen Membran zu. Diese so genannte Langzeitpotenzierung wird durch eine in Ruhe bestehende spannungsabhängige Blockade des Ionenkanals durch Mg2+-Ionen vermittelt und ist wesentliches Element synaptischer Bahnung und damit der Induktion neuronaler Plastizität (Cull-Candy und Leszkiewicz 2004). Insbesondere für Funktionssysteme wie Lernen und Gedächtnis sowie neuronale Entwicklung und Schmerzwahrnehmung spielen NMDA- Rezeptoren daher eine wichtige Rolle (Cull-Candy und Leszkiewicz 2004; Morris et al. 1986; Tsien et al. 1996). Des Weiteren kommt NMDA-Rezeptoren auch eine Funktion als Mediatoren der Homöostase der neurovaskulären Einheit zu. So konnten Vazana et al. 2016 eine Glutamat-vermittelte Erhöhung der BHS-Permeabilität durch NMDA-Rezeptor- Überaktivierung zeigen (Vazana et al. 2016; Xhima et al. 2016). Sowohl eine Überaktivierung von NMDA-Rezeptoren als auch eine zu geringe Rezeptor- aktivität kann somit eine potentiell schädigende Wirkung haben. Während eine exzitotoxi- sche Überaktivität dabei als zugrunde liegender pathophysiologischer Mechanismus im Rahmen von Epilepsien, Demenzen und ischämischen Schlaganfällen diskutiert wird, ist eine verminderte Aktivität mit psychiatrischen Störungen und Schizophrenie assoziiert (Waxman und Lynch 2005; Coyle 2006; Lau und Zukin 2007). 1.4.2 Pathologische Effekte von NMDA-Rezeptor-Autoantikörpern ZNS-reaktive AK gegen Untereinheiten des glutamatergen NMDAR werden im Zusammenhang mit verschiedenen ZNS-Pathologien beschrieben. Meist treten die AK dabei im Rahmen autoimmuner oder paraneoplastischer Prozesse auf und werden mit Anfallsleiden, Enzephalopathien sowie neurodegenerativen und neuropsychiatrischen Symptomen assoziiert (Diamond et al. 2009). Bedeutendstes Beispiel ist die von Dalmau et al. 2007 beschriebene Anti-NMDA-Rezeptor- Enzephalitis, die vornehmlich als paraneoplastisches Syndrom bei jungen Patientinnen mit ovariellen Teratomen auftritt. Patienten mit NMDAR-AK gegen die N-terminale extra- zelluläre Domäne der Rezeptor-Untereinheit NR1 entwickeln dabei eine rasch progrediente Symptomatik mit Psychosen, Gedächtnisstörungen, Sprachstörungen, epileptischen An- fällen, Vigilanzminderung und Katatonie, sowie Dyskinesien, autonome Instabilität und Hypoventilation (Dalmau et al. 2008; Dalmau et al. 2011). Die Ausprägung der Symptome korreliert mit der Höhe der AK-Titer (Hughes et al. 2010). Aufgrund der multiplen neurologischen und autonomen Defizite ist zu Beginn häufig eine intensivmedizinische Behandlung notwendig. Terratomentfernung und immunsuppressive Behandlung führen jedoch innerhalb von Wochen bis Monaten häufig zu einer Rückläufigkeit der Symptomatik (Dalmau et al. 2008; Titulaer et al. 2013). Die NMDAR-AK vermitteln ihren Effekt dabei vermutlich durch Rezeptorbindung und -internalisation, was zu einer
1 Einleitung 12 verminderten postsynaptischen Rezeptordichte und damit Störung der oben beschriebenen postsynaptischen NMDAR-Funktionen führt (Dalmau et al. 2011; Hughes et al. 2010). Daneben sind weitere mit NMDAR-AK assoziierte Erkrankungen bekannt, wie beispielsweise der bereits erwähnte neuropsychiatrische Lupus erythematodes, bei dem Patienten AK gegen die NMDAR-Untereinheit NR2 exprimieren (Diamond et al. 2009; DeGiorgio et al. 2001). Insbesondere die Prävalenz dieser AK im Liquor korreliert mit dem Auftreten neuropsychiatrischer Symptome wie Störungen von Kognition und Verhalten. Es ist jedoch derzeit noch Gegenstand aktueller Forschung inwiefern diese AK Ursache oder Folge der Erkrankung sind (Diamond et al. 2013). 1.4.3 NMDAR als Target in der Schlaganfall-Therapie NMDAR-vermittelte Exzitotoxizität stellt im Rahmen ischämischer Schlaganfälle einen wichtigen pathophysiologischen Mechanismus für neuronale Degeneration und Gewebeschädigung dar (Dirnagl et al. 1999; Cull-Candy und Leszkiewicz 2004). Sauerstoff- und Glukosemangel im ischämischen Areal verursachen einen Zusammenbruch des Membranpotentials, was einen präsynaptischen Ca2+-Einstrom mit konsekutiver Transmitterausschüttung und Glutamatakkumulation im synaptischen Spalt nach sich zieht (Katsura et al. 1994). Dies führt zu einer exzessiven Aktivierung postsynaptischer NMDAR und somit unkontrolliertem Na+-, Cl-- und Ca2+-Einstrom, Ödembildung und Calzium- vermittelter Aktivierung verschiedener Signalkaskaden. Es folgt die Initiation von inflammatorischen Prozessen, Apoptose und Peri-Infarkt-Depolarisation (Dirnagl et al. 1999). Eine Antagonisierung dieser Exzitotoxizität über eine NMDAR-Blockade stellt somit einen potentiellen Angriffspunkt in der Schlaganfall-Therapie dar (Kalia et al. 2008). Viel- versprechende präklinische Ergebnisse zum therapeutischen Einsatz von NMDAR- Antagonisten konnten in randomisierten, kontrollierten klinischen Studien jedoch nicht repliziert werden (Muir 2006; Kalia et al. 2008). Als Ursache wird unter anderem die Kürze des therapeutischen Zeitfensters diskutiert, in dem durch NMDAR-Antagonisierung eine Verminderung der Exzitotoxizität zu erwarten wäre und welches im klinischen Alltag nicht einzuhalten ist (Ikonomidou und Turski 2002). Ein sofort verfügbarer Antagonist könnte dieses Problem lösen. 1.4.4 Arbeiten zu NMDAR-ABs in der Arbeitsgruppe Das dieser Dissertation zugrundeliegende Projekt ist im Kontext mit anderen Arbeiten der AG Ehrenreich zur Rolle von NMDAR-AK im Rahmen neuropsychiatrischer und neurodegenerativer Erkrankungen zu sehen, die auch im Review von Ehrenreich 2017 zusammengefasst dargestellt sind. Hammer et al. 2014a führten eine systematische Untersuchung der Seroprävalenz von NMDAR-AK im Serum von 2817 gesunden und neuropsychiatrisch erkrankten Personen
1 Einleitung 13 durch. Dabei konnten in 10,5 % der getesteten Probanden NMDAR-AK gegen die NR1 Untereinheit detektiert werden, wobei zwischen Patienten und gesunden Kontrollen kein Unterschied in Seroprävalenz, AK-Titer oder In-vitro-Funktionalität der AK feststellbar war. Dieses überraschende Ergebnis führte zu der Hypothese, dass die Integrität der BHS von zentraler Relevanz für die Pathogenität dieser AK ist, wofür sich in Tierversuchen mit ApoE-Knockout-Mäusen und anhand einer stärkeren psychiatrischen Symptomatik seropositiver Schizophrenie-Patienten mit anamnestisch temporärer BHS-Störung (Geburtskomplikationen, Schädel-Hirn-Traumata) weitere Indizien fanden. Als prä- dispositionierende Faktoren für eine NMDAR-NR1-AK Seropositivität konnten abgelaufene Infektionen mit Influenza A oder B, sowie der SNP rs524991 identifiziert werden (Hammer et al. 2014a). Dahm et al. 2014 konnten im Rahmen eines erweiterten Screenings auf 25 ZNS-reaktive AK das zuvor erzielte Ergebnis der vergleichbaren AK-Seroprävalenz in gesunden und erkrankten Personen in einer Stichprobe von 4236 Individuen replizieren. NMDAR-NR1- AK waren dabei wiederum in durchschnittlich 10 % der Patienten nachweisbar und zeigten einen altersabhängigen Anstieg auf bis zu 20 %. Die hohe AK-Seroprävalenz ließ jedoch die Frage offen, inwiefern bei NMDAR-NR1-AK- seropositiven Personen AK auch im Liquor nachweisbar sind. Castillo-Gomez et al. 2016a adressierten diese Frage und stellten fest, dass auch bei seropositiven Individuen mit gestörter BHS ein derartiger Nachweis meist nicht möglich ist. Stattdessen ließ sich im Tierversuch zeigen, dass eine spezifische Bindung der ZNS-reaktiven AK im Gehirn erfolgt, weshalb diese im Liquor nicht mehr detektiert werden können. Im Gegensatz dazu werden AK ohne Bindungsstellen im Gehirn, im Versuch durch mit Green-Fluorescent- Protein (GFP) markierte Nonsense-AK simuliert, komplett drainiert und sind anschließend auch im Liquor nachweisbar (Castillo-Gomez et al. 2016a). Weitere Versuche zu den Charakteristika und der Funktionalität verschiedener NMDA- NR1-AK zeigten, dass alle zirkulierenden AK unabhängig von Immunglobulinklasse und Krankheitsstatus des Probanden (neuropsychiatrisch erkrankt oder gesunde Kontroll- person) in Abhängigkeit von der BHS-Integrität eine potentiell pathogene Wirkung entfalten können, indem sie eine NMDAR-Internalisierung provozieren und so zur Reduktion glutamaterger Signalübertragung führen (Castillo-Gomez et al. 2016b).
Sie können auch lesen

















































