BUNSENMAGAZIN 1/2010 - Leitartikel Der Bologna-Express droht Unterricht Mössbauer Spectroscopy - Portal
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

1/2010
BBPCAX 101 (8) 1083-1196 (1998)
ISSN 0005 – 9021
No. 1 – JANUAR 2010
BUNSENMAGAZIN
Leitartikel
Der Bologna-Express droht
zu entgleisen S. 1
Unterricht
Mössbauer Spectroscopy S. 4
Über elektrochemische Zellen S. 23
Aspekte
Kernenergie S. 29
IMPRESSUM Herausgeber: Geschäftsführer der Deutschen
Bunsen-Gesellschaft
Vorstand der Deutschen
Bunsen-Gesellschaft Andreas Förster
Bunsen-Magazin Wolfgang von Rybinski Theodor-Heuss-Allee 25
Heft 1 Jahrgang 12 Katharina Kohse-Höinghaus D-60486 Frankfurt
Wolfgang Grünbein Tel.: 069 / 75 64 620
Fax: 069 / 75 64 622
E-Mail: foerster@bunsen.de
Schriftleiter:
Peter C. Schmidt / Rolf Schäfer
Eduard-Zintl-Institut für Anorganische
und Physikalische Chemie Technische Herstellung:
Technische Universität Darmstadt VMK-Druckerei GmbH
Petersenstr. 20 Faberstraße 17
D-64287 Darmstadt D-67590 Monsheim
Tel.: 06151 / 16 27 07 oder 16 24 98 Tel.: 06243 / 909 - 110
Fax: 06151 / 16 60 15 Fax: 06243 / 909 - 100
E-Mail: bunsenmagazin@bunsen.de E-Mail: info@vmk-druckerei.de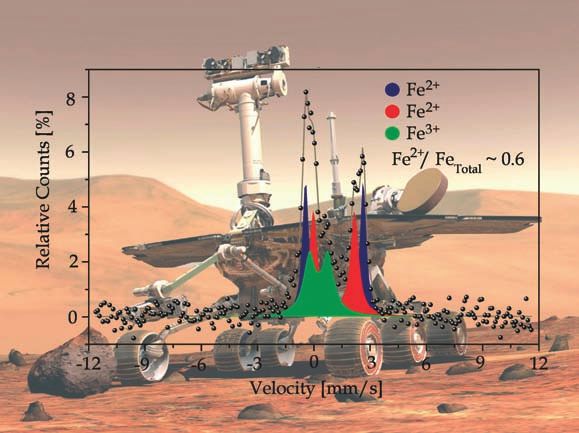
LEITARTIKEL
DEUTSCHE BUNSEN-GESELLSCHAFT
Hans-Jürgen Bär
DER BOLOGNA-EXPRESS DROHT
ZU ENTGLEISEN
Pünktlich zum 10-Jährigen Jubiläum Bereitschaft zu radikalen Änderungen wuchs. Kurzum, es wur-
der Bologna-Erklärung befinden sich de eine Kommission eingesetzt (damals noch LuSt-Ausschuss
die Studierenden an deutschen Univer- genannt, also Lehr- und Studienausschuss – welch euphemis-
sitäten im Streik. Wesentlicher Grund tisches Akronym) und wir machten uns an die Arbeit, unseren
ist eine heftige Kritik an den neuen Ba- neuen Studiengang zu gestalten. Ich musste dabei lernen,
chelor- und Master-Studiengängen. Die- dass man um Kreditpunkte wunderbar feilschen kann, sich of-
se sind in Deutschland inzwischen fast fensichtlich gar die Wertschätzung des Fachgebiets in der Zahl
flächendeckend eingeführt, die großen der ihm zugewiesenen Kreditpunkte bemisst. Fachübergrei-
Bollwerke des Widerstands sind ver- fende Veranstaltungen tun sich da schwer, schließlich müssen
schwunden und selbst ein kleines galli- die Kreditpunkte ja irgendwo herkommen. Wir haben gelernt,
sches Dorf ist nirgends mehr zu sehen. dass man ein 5-Jähriges Diplomstudium nicht in einen 3-Jäh-
Was hat uns dieser Bologna-Prozess in rigen Bachelor zwängen kann, ohne substantiell inhaltlich zu
der Chemie gebracht? Der Abschluss streichen. Auch eine kühne Kreditpunkte-Arithmetik lindert
„Diplom-Chemiker(in)“ ist abgeschafft, ebenso der Titel „Dip- dieses Problem nicht, schließlich wachen die Studierenden
lom-Ingenieur(in) in Chemie“ (was waren wir in Darmstadt stolz sehr genau darüber, dass der zulässige „work-load“ nicht über-
auf diesen Titel). Die Staatsexamens-Studiengänge werden an schritten wird. Schummeln geht hier nicht, alles muss ja fein
die Erfordernisse des Bologna-Prozesses „angepasst“, d.h. die säuberlich in Modulbeschreibungen festgehalten werden. So
Chemielehrerausbildung wird (mal wieder) reformiert. manches Praktikum wurde also zusammengestrichen, man-
Dies ist der Versuch einer Bestandsaufnahme. Sehr subjektiv che Vorlesung und Übung entfiel. Trotz allem, am Ende fand
und persönlich, daher keinesfalls repräsentativ, denn zu un- sich ein guter Kompromiss. Es entstand neben viel Papier ein
terschiedlich sind die Chemie-Fachbereiche und –Institute an ausgewogener Studiengang, von dem Lehrende wie Studieren-
deutschen Universitäten und Hochschulen. Vielleicht findet sich de überzeugt waren, der moderne Elemente (Projektunterricht)
so mancher im Gesagten wieder, vielleicht gibt es aber auch enthielt und eine überschaubare Zahl von Prüfungsereignissen
heftigen Widerspruch. beinhaltete. Es ist ja nicht so, als hätten wir noch nie einen Stu-
diengang konzipiert oder reformiert.
Als wir am Fachbereich Chemie der Technischen Universität Dann kam die Akkreditierungsagentur. Die Ernüchterung war
Darmstadt von den Plänen einer umfassenden Studienreform ungefähr so groß wie die Rechnung derselbigen. Der reformier-
hörten, war die Begeisterung zunächst gedämpft. Allerdings, te Studiengang wurde korrigiert, die Vorgaben der Akkreditie-
eine Reform des Diplomstudiengangs war dringend angesagt. rungskommission wurden umgesetzt, zum Teil mit großem Mur-
Alter Ballast sollte abgeworfen werden (Trennungsgänge im ren der Lehrenden und Bedenken der Studierenden. Module
analytischen Praktikum!), moderne Forschungsthemen ver- wurden verkleinert, die Zahl der Prüfungsereignisse wuchs. Die
stärkt in die Lehre integriert werden. Fachübergreifend sollte Forderung nach einem disjunkten Themenkatalog erschwerte
es sein, der Entwicklung in vielen Bereichen der Chemie Rech- die vertiefende Wiederholung des Lernstoffes genauso wie das
nung tragend, wo interdisziplinäre Forschung die traditionelle Verbot, einmal Geprüftes wiederholt in anderen Modulen ab-
Aufteilung der Chemie in die Grundlagenfächer aufbricht. Die zuprüfen. Wie soll man sich vernünftig über Zustandssummen
Vergleichbarkeit von Studienleistungen innerhalb Europas war unterhalten, wenn Thermodynamik und Quantenchemie nicht
schwierig, hier bot das ECTS (European Credit Transfer System) mehr Gegenstand der Modulbeschreibung sind? Hier war also
neue Möglichkeiten. Vielleicht könnte dadurch ein Auslands- Kreativität gefragt! Am Ende stand der Studiengang (der hatte
aufenthalt erleichtert und die Studierendenmobilität gefördert nur noch wenig mit unserem ersten Entwurf zu tun), er wurde
werden. Diplomprüfungen würden durch studienbegleitende akkreditiert und die ersten Studierenden schrieben sich ein.
Prüfungen ersetzt werden, was von den Studierenden zu- Erst sehr zögerlich (wir hatten als Alternative im ersten Jahr
nächst heftig begrüßt wurde. Die Gelegenheit war günstig, da noch das Diplom), dann blieb ihnen keine andere Wahl (das
die Professorenschaft sich zusehends verjüngte und somit die Diplom war endgültig abgeschafft).
Dr. Hans-Jürgen Bär
Fachbereich Chemie, TU Darmstadt
Petersenstraße 20, D-64287 Darmstadt
Telefon: +49/6151/164095,
E-Mail: h.baer@theo.chemie.tu-darmstadt.de
1
LEITARTIKEL
BUNSEN-MAGAZIN · 12. JAHRGANG · 1/2010
Wie ist die Situation heute, 4 Jahre nach der Einführung des durchgedrückt. Auf ihrer Homepage [1] findet sich ein hochinte-
Bachelors? Die Studierendenzahlen steigen (zum Glück) weiter ressantes Statement zur aktuellen Situation mit der Bemerkung
an, aber dies ist sicher nicht dem Bachelor geschuldet. Die No- „Die Umsetzung der Bologna-Vereinbarung aber ist an zu vielen
tenfülle führt meist zu einer mittleren Gesamtnote und selbst Hochschulen nicht gut gelungen“. Dies ist auch der Tenor der
beste Bachelor-Studierende tun sich schwer, einen sehr guten derzeitigen Bildungsministerin, die von „handwerklichen Feh-
Abschluss zu erreichen. Bei der Übernahme in ein Master-Pro- lern“ bei der Einführung spricht. Klar ausgedrückt: die Politik hat
gramm oder bei der Stipendienvergabe muss dies in Zukunft das Richtige gewollt, aber die Universitäten haben es verbockt.
unbedingt berücksichtigt werden. Seitens der Politik wird die
Einführung der neuen Studiengänge auch mit der Forderung Trotz Studierenden-Streiks und Politikerschelte sollten wir aber
nach einer Reduzierung der Abbrecherquoten verbunden. Bei mit Optimismus in die Zukunft blicken. Für die Chemie ist die
einer zunehmenden Stoffdichte kann dies letztlich trotz aller Einführung gestufter Studiengänge durchaus vorteilhaft. Ei-
verpflichtenden Tutoren- und Mentorenprogramme nebst Stu- ner breiten Ausbildung im Bachelor steht eine Spezialisierung
dienberatungen nur durch ein Absenken des Prüfungsniveaus im Master gegenüber, der auf die Promotion vorbereitet. Ein
erreicht werden, was bewusst oder unbewusst auch passiert. umfangreicher Wahlpflichtbereich sollte es Studierenden er-
Wenn ich aktuelle Klausuren und Übungsblätter aus der Physi- möglichen, über den Tellerrand hinauszusehen und andere
kalischen Chemie mit denen aus den 80er Jahren vergleiche, Fächer (von der Biologie über den Maschinenbau bis hin zur
kommen mir die Tränen. Betriebswirtschaftslehre) gewinnbringend in den Studiengang
Studierende beklagen – nicht nur in der Chemie – die „Lern- zu integrieren. Bei den Praktika müssen Abstriche gemacht
bulemie“. Wissen in sich reinprügeln, dann prüfen lassen und werden und wir müssen auch akzeptieren, dass die drei „Kern-
alles wieder vergessen. Schließlich wird es ja nicht nochmals fächer“ Anorganische, Organische und Physikalische Chemie
abgefragt, der Akkreditierung sei Dank. Das Studium ist inzwi- nicht mehr die Eckpfeiler der Chemieausbildung sind. Was in
schen vollständig verschult. Prüfungsordnungen sind so kompli- der Forschung aufgebrochen wird, muss sich letztlich auch im
ziert, dass eigens Studienkoordinatoren über deren Einhaltung Unterricht (und in den Fachbereichsstrukturen!) widerspiegeln.
wachen müssen. Dozenten haben häufig bereits den Über- Nutzen wir die neuen Möglichkeiten der Evaluierung und des
blick über die bürokratischen Prüfungsabwicklungen verloren. Mentorensystems zu einer steten Verbesserung der Lehre. Die
Die Kritik von Lehrenden und Studierenden ist immens und hat Zeit ist günstig, im Zuge der Studierendenproteste Prüfungs-
inzwischen dazu geführt, dass die Reform wieder reformiert bürokratie abzubauen und Ecken und Kanten der Studien- und
wird. Also wird korrigiert (und versprochen), aber mit mageren Prüfungsordnungen zu schleifen, so dass die Studiengänge
Ergebnissen: die Kultusminister der Länder haben die Reform auch studierbar bleiben (oder werden).
der Reform erst mal auf Eis gelegt, schließlich müssen die föde- Auf eine Sache müssen wir aber sorgfältig achten: das Niveau
ralen Interessen sorgfältig abgewogen werden. Als Trostpflaster der Chemie-Ausbildung darf nicht weiter sinken. Allzu tole-
soll immerhin das Bafög erhöht werden. Seltsam ist – und das rantes Öffnen der Studiengänge und gleichzeitige Vorgaben
sei hier ausdrücklich angemerkt – dass sowohl die Universitä- über Absolventenquoten sind hier nicht zielführend. Stattdes-
ten als auch die Industrie mit Kritik an der momentanen Situ- sen sollte das Werkzeug des Eignungsfeststellungsverfahrens
ation nicht sparen, sie bei der Einführung der neuen Studien- stärker genutzt werden. Das sind wir dem Forschungsstandort
gänge aber überraschend schweigsam waren. Die Umsetzung Deutschland, vor allem aber unseren Studierenden, schuldig.
des Bologna-Prozesses wurde übrigens seinerzeit von einer Schließlich sind die Studierenden von heute die Doktorandin-
Bildungsministerin gegen die Zuständigkeit der Bundesländer nen und Doktoranden von morgen.
[1] http://www.edelgard-bulmahn.de/aktuelles/nachrichten/2009/100285.php
2
INHALTSVERZEICHNIS
DEUTSCHE BUNSEN-GESELLSCHAFT
Leitartikel
Hans-Jürgen Bär
Der Bologna-Express droht zu entgleisen 1
Unterricht
Philipp Gütlich und Christian Schröder
Mössbauer Spectroscopy 4
Bruno Boddenberg
Über Galvanipotentiale und –spannungen, Elektrodenpotentiale
und die EMK einer elektrochemischen Zelle 23
Leserbrief
Leserbriefe zu unserer Serie über „Chemie und Energie“ 27
Aspekte
Ulrich Schindewolf und Joachim Hornke
Kernenergie 29
Tagungen
Katharina Al-Shamery
Aus den Hexenküchen der Materialwissenschaften 43
Nachrichten
Michael Buback zum 65. Geburtstag 45
Personalia 47
Veranstaltungen/Events 48
Ausschreibung/Ankündigung 49
Physikalische Chemie
Inhalt Heft 10 - 11 (2009) 52
Zum Titelbild
“The image on the front cover shows a history-making Mössbauer
spectrum, the first Mössbauer spectrum obtained of the surface of
Mars. The spectrum was obtained in January 2004 on sol 14 after
the landing of the NASA Mars Exploration Rover Spirit. One sol is a
martian day, which is slightly longer than the 24 hrs of an Earth day.
The background shows an artist’s conception of a Mars Exploration
Rover (source of the background image: NASA/JPL/Cornell).” Siehe
Artikel von Philipp Gütlich und Christian Schröder, Seite 4.
3
UNTERRICHT
BUNSEN-MAGAZIN · 12. JAHRGANG · 1/2010
Philipp Gütlich and Christian Schröder
MÖSSBAUER SPECTROSCOPY
INTRODUCTION trial applications. Remarkable progress has also taken place
in instrumentation and methodology. Most spectacular, for
Nearly fifty years ago Rudolf L. Mössbauer, whilst working instance, is the miniaturisation of a laboratory spectrometer
on his doctoral thesis under Professor Maier-Leibnitz in Munich by scaling down by a factor of ca. one hundred to a device,
and Heidelberg, discovered the recoilless nuclear resonance known as MIMOS, which currently operates on the surface of
absorption of g-rays which became known as the Mössbauer the planet Mars as well as in many mobile analytical studies
Effect [1-3]. The phenomenon rapidly developed to a new spec- on Earth (see below). The second most remarkable develop-
troscopic technique which now bears his name. Mössbauer ment began in the mid eighties with the experimental proof by
spectroscopy has made valuable contributions to the physical-, Gerdau et al. [4] that nuclear resonance fluorescence is also
chemical-, biological- and geo-sciences. possible with synchrotron radiation instead of a radioactive ra-
Mössbauer’s discovery that γ-ray emission and absorption diation source. Two methods have emerged thereof: Nuclear
can occur in a recoil-free fashion might have seemed at first Forward Scattering (NFS) for the measurement of hyperfine in-
glance to be no more than just an interesting new phenomenon. teraction, and Nuclear Inelastic Scattering (NIS) of synchrotron
However, as soon as it became generally realised that the Möss- radiation for the measurement of phonon spectra of local vi-
bauer resonance line is extremely narrow and allows hyperfine brational modes nearby the Mössbauer probe nucleus. These
interactions to be resolved and evaluated in a rather straight- two methods will not be covered further in this text, however.
forward way, this handy new method created an avalanche of We refer the interested reader to available reviews (e.g., [5]).
research activity. Within a few years nearly all disciplines in the Over the years, many introductory texts about the principles
natural sciences enjoyed a boom in the application of Möss- and applications of Mössbauer spectroscopy have been writ-
bauer spectroscopy. Some journals were swamped to such an ten. We list those that go beyond the scope of this text [6-22].
extent that editorials were written to limit the publication of
Mössbauer results. Rudolf Mössbauer’s concluding remark con-
cerning the effect that bears his name in his Nobel Laureate BASIC PRINCIPLES
speech of December 1961 has proved to be correct and has re-
tained its significance to the present day; it can also be regarded Figure 1 shows the periodic table of the elements where
as a prognosis for the future: “We may therefore hope that this the elements (more than 40) for which the Mössbauer effect
young branch of physics stands only at its threshold, and that it
will be developed in the future, not only to extend the application
of existing knowledge but to make possible new advances in the
exciting world of unknown phenomena and effects.”
Mössbauer spectroscopy has become an elegant tool for
the study of electronic structure, bonding properties, mo-
lecular symmetry, magnetic behaviour and phase transitions
in solid state. Even substances that a priori do not contain a
Mössbauer active nuclide have been extensively studied re-
garding, e.g. structural phase transitions, by doping the sample
with small amounts of a Mössbauer probe nuclide (e.g. 57Fe).
Also, although the Mössbauer effect is observable only in solid
material, it is well established that Mössbauer spectra may
be recorded of frozen solutions in order to study, for example,
electron transfer and ligand exchange reactions. Figure 1. Periodic table of the elements where the elements for which the
Not only has the applicability of Mössbauer spectroscopy Mössbauer effect has been observed are highlighted. Colours represent ele-
ment categories: Alkali metals are yellow, Fe and other transition metals are
enormously spread in the characterisation of materials in the blue, for example (Source: Mössbauer Effect Data Centre;
broadest sense, from fundamental research to practical indus- http://orgs.unca.edu/medc/Resources.html).
Prof. Dr. Philipp Gütlich Dr. Christian Schröder
Institut für Anorganische Chemie und Analytische Chemie, Universität Bayreuth und
Johannes Gutenberg-Universität, Eberhard Karls Universität Tübingen
Staudinger Weg 9, 55128 Mainz, Germany Sigwartstr. 10, 72076 Tübingen, Germany
Phone: +49-6131-39-22373, Fax: +49-6131-39-22990 Phone: +49-7071-29-78924, Fax: +49-7071-29-5059
E-Mail: guetlich@uni-mainz.de E-Mail: christian.schroeder@ifg.uni-tuebingen.de
4
UNTERRICHT
DEUTSCHE BUNSEN-GESELLSCHAFT
has been observed are marked. The Mössbauer effect has state. In the solid state, crystalline or non-crystalline, recoilless
been detected with a total of nearly 90 γ-ray transitions in 72 emission and absorption of g-quanta is possible, and the essen-
isotopes of 42 different elements. Due to several criteria (suit- tially unshifted transition lines can (at least partially) overlap
able lifetime of nuclear excited state, transition energy, easy and nuclear resonance absorption can be observed. The rea-
accessibility and handling) only ca. twenty elements can be son is that due to the much larger mass M of a solid particle as
studied by Mössbauer spectroscopy, e.g. iron, tin, antimony, compared to that of an atom or a molecule, the linear momen-
tellurium, iodine, gold, nickel, ruthenium, iridium, tungsten, tum created by emission and absorption of a g-quantum practi-
krypton, xenon, many of the rare earth elements, and neptu- cally vanishes according to equation 2. The recoil energy caused
nium. The most prominent “Mössbauer nuclide” is 57Fe. More by an emitting and absorbing atom, which is tightly bound in the
than 90 % of the more than 50 000 publications which have lattice, is mostly transferred to the lattice vibrational system.
appeared so far refer to 57Fe spectroscopy. For pedagogical There is a certain probability f (known as Debye-Waller factor or
reasons the following paragraphs refer to the most prominent Lamb-Mössbauer factor) that no lattice excitation (zero-phonon
Mössbauer active nuclide 57Fe. processes) takes place during γ-emission or γ-absorption. The
f-factor denotes the fraction of nuclear transitions which occur
without recoil. Only for this fraction is the Mössbauer effect ob-
THE MÖSSBAUER EFFECT servable. Within the Debye model for solids, f increases with
decreasing transition energy Eγ, with decreasing temperature,
The Mössbauer effect is the recoilless nuclear resonance ab- and with increasing Debye temperature θD = hωD/2πkB (ωD =
sorption and emission of g-rays, similar to the acoustic reso- vibrational frequency of Debye oscillator, kB = Boltzmann fac-
nance between two tuning forks with the same frequency fs = fr tor). The Debye temperature θD is a measure of the strength of
for sender (s) and receiver (r). A nucleus with Z protons and N the bonds between the Mössbauer atom and the lattice. It is
neutrons in an excited state of energy Ee undergoes transition high (greater than room temperature) for metallic materials and
to the ground state of energy Eg by emitting a γ-quantum of low (lower than room temperature) for soft compounds such as
energy Ee – Eg. The γ-quantum may be absorbed by a nucleus metalorganic compounds.
of the same kind (same Z and N) in its ground state, whereby
transition to the excited state of energy Ee takes place (reso-
nance absorption). The subsequent transition to the ground THE MÖSSBAUER SPECTROMETER
state emits a conversion electron e- (with nearly ten times
higher probability than γ-emission) or a γ-quantum (resonance The main components of a Mössbauer spectrometer are, in the
fluorescence). simplest terms, the source emitting the characteristic γ-rays
An excited state (nuclear or electronic) of mean lifetime (called Mössbauer source), the sample or absorber to be ana-
τ can never be assigned a sharp energy value, but only a lyzed (Mössbauer absorber), and the detector system (Figure 2).
value within the energy range ΔE, which correlates with the The source, for 57Fe spectroscopy one uses generally commer-
uncertainty in time Δt via the Heisenberg Uncertainty Princi- cially available 57Co/Rh, is mounted on the shaft of a vibrator.
ple: ΔEΔt ≤ ħ. Weisskopf and Wigner have shown that in gen- Source and absorber are moved relative to each other with the
eral Г · τ = ħ, where Г is the natural line width and ħ = h/2π is Doppler velocity
Planck’s constant. The mean lifetime τ determines the width of
the resonance lines (G · τ = ħ) and is related to the half-life t1/2 (3) n = c(G / Eg).
by the relation τ = ln2 ∙ t1/2. According to Weisskopf and Wigner
the distribution of energies about the most probable energy E0
(= transition probability as function of transition energy E) is
given by the Breit-Wigner (or Lorentzian) formula:
(* / 2) 2
(1) I (E) .
( E E 0 ) 2 (* / 2) 2
Resonance absorption is observable only if the emission and
absorption lines overlap sufficiently. By emission or absorption
of γ-quanta with energy Eγ in a free atom or molecule (gas,
liquid), where the transition energy Eγ is slightly changed com-
pared to E0 of a ‘naked’ nucleus due to interactions between
protons and electrons penetrating the nuclear field, the atom
(molecule) of mass m suffers a recoil effect with energy ER
given by the equation
(2) ER = Eg2 / 2mc2,
which is much larger (5-6 orders of magnitude) than the natural
line width G. No resonance is possible between free atoms or
molecules. The Mössbauer effect therefore cannot be observed
for freely moving atoms or molecules, i.e. in gaseous or liquid Figure 2. Scheme of a Mössbauer spectrometer.
5
UNTERRICHT
BUNSEN-MAGAZIN · 12. JAHRGANG · 1/2010
For 57Fe, Γ = 4.7·10-9 eV, Eg 14400 eV, and v = 0.096 mm s-1. Mössbauer spectroscopy. The nuclear spin quantum numbers
While the source is generally kept at room temperature, the ab- of the excited state (14.4 keV) and the ground state are I = 3/2
sorber (sample under study) may be thermostated in a cryostat and I = 1/2, respectively. The internal conversion coefficient α
for cooling down to liquid nitrogen or liquid helium temperatures, (= the number of ejected K-shell electrons for each γ-quantum
or for controlled heating in an oven. The γ-rays are detected by a interacting with the K-shell) is 9.7.
scintillation counter, gas proportional counter or a semi-conduc- Recoilless resonant absorption is necessary for maximum
tor detector. The pulses from the detector are amplified and pass overlap of the emission line (E) and absorption line (A). Only
through a discriminator, where most of the non-resonant back- for identical materials, e.g. 57Co diffused into stainless steel as
ground radiation is rejected, and finally are fed into the open a source and stainless steel containing 57Fe (to 2.2 % natural
channel address of a multi-channel analyser (e.g. a computer) abundance) as absorber at the same temperature can total
with several hundred channels, which is synchronised with the overlap be expected. If, however, the source and the absorber
vibrator making use of the so-called feed-back control system. consist of different materials, which is usually the case when
A constant frequency clock synchronises a voltage waveform studying an iron-containing sample as absorber with a 57Co/Rh
(usually triangular yielding a linear Doppler velocity scale) which source, the resonance effect may be perturbed due to electric
serves as a reference signal to the servo-amplifier controlling the and magnetic hyperfine interactions between the nuclei and
electro-mechanical vibrator. The difference between the moni- electric and magnetic fields set up by electrons interacting with
tored signal and the reference signal is amplified and drives the the nuclei (see below). Such hyperfine interactions not only
vibrator at the same frequency (typically 50 s-1) as the channel shift, but may also split degenerate nuclear levels resulting in
address advances. Each channel corresponds to a certain rela- several transition lines. The Mössbauer source is always pre-
tive velocity and is held open for a fixed time interval depending pared such that it emits a single transition line E and we as-
on the frequency and number of channels used. The incoming sume, for the sake of simplicity, that the absorber shows also
g-counts are collected in their corresponding channels during the only one transition line A. But E and A now have slightly modi-
sequential accessing, e.g. 50 times per second, until satisfac- fied transition energies Eγ; the perturbation energies are of the
tory resolution is reached. The measured Mössbauer spectrum order of 10-8 eV (comparable to the natural line width Γ), which
is analysed using special least squares fitting programs. shifts the transition lines away from each other such that the
Radioactive 57Co with 270 days half-life, which may be overlap decreases or disappears entirely. Perfect overlap can
generated in a cyclotron and diffused into a noble metal like be restored again by making use of the Doppler effect, i.e. by
rhodium, serves as the g-radiation source for 57Fe Mössbau- moving the absorber (generally kept fixed) and the source (gen-
er spectroscopy. The isotope 57Co decays by electron capture erally mounted on a vibrator) relative to each other (Figure 4).
(EC from K-shell, thereby reducing the proton number from 27 In the case of 57Fe spectroscopy, Doppler velocities of up to a
to 26 corresponding to 57Fe; Figure 3) and initially populates few mm s-1 are sufficient to make up for the perturbing hyper-
the 136 keV nuclear level of 57Fe with nuclear spin quantum
number I = 5/2. This excited state decays after ca. 10 ns and
populates, with 85 % probability, the 14.4 keV level by emitting
122 keV γ-quanta, with 15 % probability the 136 keV level de-
cays directly to the ground state of 57Fe. The 14.4 keV nuclear
state has a half-life of ca. 100 ns. Both the half-life and the
emitted γ-quanta of 14.4 keV energy are ideally suited for 57Fe
Figure 4. In a Mössbauer experiment, perfect overlap between emission (E)
and absorption (A) lines can be restored by making use of the Doppler effect,
i.e. by moving the source (generally mounted on a vibrator) and the absorber
(generally kept fixed) relative to each other.
fine interaction energies and bring emission and absorption
lines to perfect overlap, i.e. resonance. The hyperfine interac-
tion energy ε (of interest) is correlated with the Doppler velocity
v via ε = (v/c) Eγ and can be measured in this way. The plot of
the relative transmission of the g-radiation as a function of the
Doppler velocity v is called the Mössbauer spectrum.
In order to measure the absorption, Mössbauer samples
Figure 3. Simplified nuclear decay scheme of 57Co for 57Fe Mössbauer reso- need to be prepared sufficiently thin, e.g. as powders or thin
nance. slices, to let radiation pass through to the detector (transmis-
6
UNTERRICHT
DEUTSCHE BUNSEN-GESELLSCHAFT
sion geometry). Alternatively, one may place the detector on the than that in the ground state: Re ≠ Rg. If the source and ab-
same side of the absorber as the source (backscattering geom- sorber materials are different, the electronic densities set up
etry; Figure 5) and measure, after de-excitation, the re-emitted by all s-electrons (1s, 2s, 3s, etc.) of the electronic shells are
γ-radiation or other means of de-excitation such as electrons or different at the nuclei of the source and the absorber: ρS ≠ ρA.
X-rays (Figure 6; [23]). This is possible because the time a chan- The result is that the electric monopole interactions (Coulomb
nel address stays open in the multi-channel analyser is much interactions) are different in the source and the absorber and
longer than the lifetime of the excited state. One can measure therefore affect the nuclear ground and excited state levels to
electrons or X-rays of distinct energies and thus probe the ab- a different extent. This leads to the measured isomer shift δ as
sorber to different depths. In backscattering geometry, the ab- sketched in Figure 7. The fictitious energy levels of the ground
sorber does not have to be prepared to be sufficiently thin. and excited states of a bare nucleus (no surrounding electrons)
are perturbed and shifted by electric monopole interactions.
The shifts in the ground and excited states differ because of
the different nuclear radii in the two states, which cause dif-
ferent Coulomb interactions. The energy differences ES and EA
in the source and absorber also differ because of the different
electron densities in the source and absorber material.
Figure 5. Schematics of a Mössbauer experiment in either transmission geo-
metry (the spectrum shows absorption lines) or in backscattering geometry
(the spectrum shows emission lines).
Figure 7. Illustration of the changes in the energy levels between source (S)
and absorber (A) as a result of different s-electron densities at the S and A
nuclei and the manifestation of the resulting isomer shift δ in the Mössbauer
spectrum.
The energy differences ES and EA cannot be measured in-
dividually, a Mössbauer experiment measures only the differ-
ence of the transition energies δ = EA – ES, which is the isomer
shift. This shift appears in the spectrum as the difference be-
tween the position of the barycentre of the resonance signal
and zero Doppler velocity. The isomer shift is given by the fol-
lowing expression:
(4) d = EA – ES = C(rA – rS) (Re2 – Rg2),
Figure 6. The different resonant excitation and de-excitations modes for 57Fe where C = (2/3) πZe2 (the dielectric constant was taken to
(Figure modified from [23]). be 1). The Doppler velocity vD necessary to restore resonance
is then given by
HYPERFINE INTERACTIONS AND MÖSSBAUER c
(5) vD G.
PARAMETERS EJ
The isomer shift depends directly on the s-electron densi-
ELECTRIC MONOPOLE INTERACTION – ISOMER SHIFT ties (as sum of contributions from all s-electron shells), but
may be influenced indirectly via shielding effects of p-, d- and
Electric monopole interaction is the Coulomb interaction f-electrons, which are not capable (if neglecting relativistic ef-
between protons of the nucleus and electrons (mainly s-elec- fects) of penetrating the nuclear field. Results from Hartree-
trons) penetrating the nuclear field. The observable Möss- Fock calculations of the contributions of s-orbitals to the total
bauer parameter is the isomer shift δ. Isomer shift values give electron density at the iron nucleus as a function of oxidation
information on the oxidation state, spin state, and bonding state and configuration have shown that (a) nominally the larg-
properties such as covalency and electronegativity. est contributions originate from the filled 1s- and 2s-shells (1s
In a typical Mössbauer experiment, the source (S) material ~104 au-3, 2s ~103 au-3, 3s ~102 au-3), and (b) significant chang-
(e.g. 57Co embedded in Rh metal) is generally different from es in the electron densities arise from the noticeably different
the absorber (A) material under study. The nuclear radius in contributions from the 3s shell populations due to different
the excited state is different (in the case of 57Fe it is smaller) shielding effects of 3dn configurations (Table 1 and Figure 8
7
UNTERRICHT
BUNSEN-MAGAZIN · 12. JAHRGANG · 1/2010
Table 1. Contributions of ns orbitals to the total electron density at the Fe nucleus (in au-3) as a function of oxidation state and configuration (from spin-
averaged Hartree-Fock calculation and large Uncontracted Gaussian Basis set) [24].
Fe0 Fe+ Fe2+ Fe3+
s2d6 s1d7 s0d8 s2d5 s1d6 s0d7 s2d4 s1d5 s0d6 s2d3 s1d4 s0d5
1s 10689.72 10690.01 10690.18 10689.37 10689.77 10690.02 10688.86 10689.43 10689.79 10688.24 10688.92 10689.45
2s 981.99 982.04 982.19 981.99 981.92 982.00 982.19 981.86 981.85 982.63 982.06 981.80
3s 134.80 132.63 131.46 137.35 134.37 132.53 141.09 136.82 134.24 145.65 140.65 136.81
4s 6.12 2.05 - 9.55 4.09 - 12.44 5.51 - 15.68 6.89 -
Figure 8. Calculated 1s-, 2s-, 3s- and 4s-electron contributions to the total elec-
tron density at the Fe nucleus (nonrelativistic B3LYP DFT calculations) [24].
[24]). The reason becomes apparent on inspecting the strongly
overlapping distribution functions of 3s and 3d electrons.
Chemical bonds between metal ion and ligands in coordi-
nation compounds can be viewed as the result of the balance
between σ-donation (s-electrons from ligands are donated into
s-orbitals of the metal) and dp-pp back donation (d-electrons
move from d-orbitals of the metal to empty p-orbitals of the Figure 9. The figure shows ranges of isomer shift values expected for diffe-
rent oxidation and spin states. The most positive isomer shift occurs with for-
ligands). Both bonding mechanisms influence the isomer shift mally iron(I) compounds with spin S = 3/2. In this case, the seven d-electrons
in the same direction, but to different extent, depending on exert a very strong shielding of the s-electrons towards the nuclear charge,
this reduces markedly the s-electron density ρA giving a strongly negative
the nature of the ligands and thus on the weight of the atomic quantity (ρA – ρS). As the nuclear factor (Re2 – Rg2) is negative for 57Fe, the
orbitals of the metal and ligands participating in the molecular measured isomer shift becomes strongly positive. At the other end of the
isomer shift scale are strongly negative values expected for formally iron(VI)
orbitals (covalency effects). This is the reason why isomer shift compounds with spin S = 1. The reason is that iron(VI) compounds have only
scales for different compounds of the same oxidation state of- two d-electrons, the shielding effect for s-electrons is very weak in this case
and the s-electron density ρA at the nucleus becomes relatively high which
ten cover a broad range of values. – multiplied by the negative nuclear factor (Re2 – Rg2) – pushes the isomer
The most valuable information derived from isomer shift shift value strongly in a negative direction. It is seen from the table that
data refers to the valence state of a Mössbauer-active atom some isomer shift regions do not overlap, e.g. iron(II) high spin compounds
with S = 2 can be easily assigned from a Mössbauer spectrum. In other cases
embedded in a solid material. Figure 9 shows ranges of iso- with more or less strong overlapping δ values unambiguous assignment to
mer shift values expected for different oxidation and spin certain oxidation and spin states may not be possible. In such cases the
quadrupole splitting parameter ΔEQ will be included in the analysis and leads
states of iron. to a conclusive characterisation in most cases.
ELECTRIC QUADRUPOLE INTERACTION – If the electric field gradient (EFG) is non-zero, for instance
QUADRUPOLE SPLITTING due to a non-cubic valence electron distribution and/or non-
cubic lattice site symmetry, electric quadrupole interaction as
Electric quadrupole interaction occurs if at least one of the visualised by the precession of the quadrupole moment vector
nuclear states involved possesses a quadrupole moment eQ about the field gradient axis sets in and splits the degener-
(which is the case for nuclear states with spin I > 1/2) and if ate I = 3/2 level into two substates with magnetic spin quan-
the electric field at the nucleus is inhomogeneous. In the case tum numbers mI = ± 3/2 and ± 1/2 (Figure 10). The energy
of 57Fe the first excited state (14.4 keV state) has spin I = 3/2 difference between the two substates ΔEQ is observed in the
and therefore also an electric quadrupole moment. spectrum as the separation between the two resonance lines.
8UNTERRICHT
DEUTSCHE BUNSEN-GESELLSCHAFT
These two resonance lines in the spectrum refer to the two the metal centre and ligands with different σ-bonding and p-
transitions between the two substates of the split excited state backbonding capability. It is understood that both sources of
and the unsplit ground state. The ground state with I = 1/2 has valence electron contributions are jointly operative and can-
no quadrupole moment and remains therefore unsplit, but still not be separated.
twofold degenerate. This degeneracy can be lifted by magnetic The electric quadrupole splitting gives information on the
dipole interaction (Zeeman effect, see below). The same holds oxidation state, the spin state and the local symmetry of the
for the two substates of the excited I = 3/2 level, which are still Mössbauer atom. Note that the isomer shift parameter δ is giv-
twofold degenerate after electric quadrupole interaction. This en by the distance of the barycentre of the quadrupole doublet
becomes apparent by looking at the expression for the quadru- from zero Doppler velocity (Figure 10).
polar interaction energies EQ derived from perturbation theory:
§ eQV zz · 2
(6) EQ ( I , m I ) > @
¨¨ 4 I (2 I 1) ¸¸ 3m I I ( I 1) (for axial symmetry).
MAGNETIC DIPOLE INTERACTION –
NUCLEAR ZEEMAN EFFECT
© ¹
For 57Fe Mössbauer spectroscopy, electric quadrupole in- The requirements for magnetic dipole interaction to be ob-
teraction in the absence of magnetic dipole interaction leads served are that (i) the nuclear states involved must possess
to a doublet, the separation of the two resonance lines giving a magnetic dipole moment and (ii) a magnetic field must be
the quadrupole interaction energy ΔEQ which is proportional present at the nucleus. A nuclear state with spin I ≥ 1/2 pos-
to the quadrupole moment eQ and the electric field gradient sesses a magnetic dipole moment m. This is the case for both
(EFG). The electric field E at the nucleus is the negative gradi- the ground state with I = 1/2 and the first excited state with
ent of the potential, –ÑV, and the electric field gradient EFG I = 3/2 of 57Fe. Magnetic dipole interaction (visualised as the
is given by the nine components Vij = (∂2V/∂i∂j) (i,j,k = x,y,z) of precession of the magnetic dipole moment vector about the
the 3×3 second rank EFG tensor. Only five of these compo- axis of the magnetic field; Figure 11) leads to splitting of the
nents are independent because of the symmetric form of the states |I, mI> into 2I+1 substates characterised by the mag-
tensor, i.e. Vij = Vji and because of Laplace’s equation which netic spin quantum numbers mI. Thus the excited state with
requires that the tensor be traceless: ÑVii = 0. In the prin- I = 3/2 is split into four, and the ground state with I = 1/2 into
cipal axes system the off-diagonal elements vanish, and for two substates. These substates are no longer degenerate. The
axial symmetry (fourfold or threefold axis of symmetry pass- energies of the sublevels are given from first-order perturba-
ing through the Mössbauer nucleus yielding Vxx = Vyy) the EFG tion theory by
is then solely given by the tensor component Vzz. For non-axial
symmetry the asymmetry parameter η = (Vxx–Vyy)/Vzz is re- (7) EM(mI) = -mBmI / I = -gNbNBmI,
quired in addition. When choosing the principal axes ordering
such that Vzz ≥ Vyy ≥ Vxx, the asymmetry parameter range be- where gN is the nuclear Landé factor and bN the nuclear
comes 0 ≤ η ≤ 1. Bohr magneton. Note that the sign of the magnetic spin quan-
In principle, there are two sources which can contribute tum numbers mI of the sublevels have a different sequence in
to the total EFG: (i) charges (or dipoles) on distant ions sur- the excited state and the ground state, this being due to the
rounding the Mössbauer atom in non-cubic symmetry, usually different signs of the magnetic moments of the two states. The
termed lattice contribution to the EFG; (ii) anisotropic (non- allowed g-transitions between the sublevels of the excited state
cubic) electron distribution in the valence shell of the Möss- and those of the ground state are given by the selection rules
bauer atom, usually called valence electron contribution to the for magnetic dipole transitions: ΔI = ±1, ΔmI = 0, ±1. The six al-
EFG. The latter comes about mainly in two ways: (i) Anisotrop- lowed transitions in the case of 57Fe are shown in Figure 11 and
ic population of the metal d-orbitals visualised in the frame Figure 12.
of simple crystal field theory with axial distortion to molecu-
lar symmetry lower than Oh (an example is given below); (ii)
anisotropic covalency effects in molecular orbitals between
Figure 10. In the case of a non-zero electric field gradient (EFG), electric
quadrupole interaction as visualised by the precession of the quadrupole
moment vector about the field gradient axis sets in and splits the degene-
rate I = 3/2 level into two substates with magnetic spin quantum numbers Figure 11. Magnetic dipole interaction (visualised as the precession of the
mI = ± 3/2 and ± 1/2. This gives rise to two transition lines with equal proba- magnetic dipole moment vector about the axis of the magnetic field) leads
bility (intensity). The energy difference between the two substates ΔEQ is ob- to splitting of the states |I, mI> into 2I+1 substates characterised by the
served in the spectrum as the separation between the two resonance lines. magnetic spin quantum numbers mI.
9UNTERRICHT
BUNSEN-MAGAZIN · 12. JAHRGANG · 1/2010
Figure 12. Typical 57Fe Mössbauer spectrum resulting from magnetic dipole in-
teraction. The splitting of the ground state and the excited state can be deter-
mined as depicted in the figure and described in the text.
The separation between the lines 2 and 4 (also between
3 and 5) refers to the magnetic dipole splitting of the ground
state. The separation between lines 5 and 6 (also between 1 Figure 13. Magnetic dipole interaction and electric quadrupole interaction
may be present in a material simultaneously (together with the electric mo-
and 2, 2 and 3, 4 and 5) refers to the magnetic dipole splitting nopole interaction which is always present). In the case of relatively weak
of the excited I = 3/2 state (Figure 12). The magnetic hyper- quadrupole interaction the nuclear sublevels |I, mI> arising from magnetic
dipole splitting are additionally shifted by the quadrupole interaction ener-
fine splitting enables one to determine the effective magnetic gies EQ(I, mI); as a result, the sublevels of the excited I = 3/2 state are no
field (size and direction) acting at the nucleus. Such a field can longer equally spaced. The shifts by EQ are upwards or downwards depending
be externally applied. But many substances can also create a on the direction of the EFG. This enables one to determine the sign of the
quadrupole splitting parameter ΔEQ. This scheme applies to Mössbauer ef-
magnetic field of their own through various mechanisms, e.g.: fect measurements containing 57Fe.
- The Fermi contact field BC arises from a net spin-up or
spin-down s-electron density at the nucleus as a conse-
quence of spin polarisation of inner filled s-shells by spin-
polarised partially filled outer shells;
- a contribution BL may arise from the orbital motion of
valence electrons with the orbital momentum quantum
number L;
- a contribution BD, called spin-dipolar field, may arise from
the total electron spin of the atom under consideration.
All contributions may be present and add to the total effec-
tive magnetic field Beff = BC + BL + BD. By applying an external
magnetic field of known size and direction one can determine
the size and the direction of the intrinsic effective magnetic
field Beff of the material under investigation.
Magnetic dipole interaction and electric quadrupole interac-
tion may be present in a material simultaneously (together with
the electric monopole interaction which is always present). The
perturbations are treated depending on their relative strengths. In
the case of relatively weak quadrupole interaction the nuclear sub-
levels |I, mI> arising from magnetic dipole splitting are additionally
shifted by the quadrupole interaction energies EQ(I, mI); as a result,
the sublevels of the excited I = 3/2 state are no longer equally
spaced. The shifts by EQ are upwards or downwards depending on
the direction of the EFG. This enables one to determine the sign of
the quadrupole splitting parameter ΔEQ (Figure 13).
SELECTED APPLICATIONS Figure 14. Mössbauer spectra of three selected iron(II) compounds. The
ferrous sulphate A, better formulated as [Fe(H2O)6]2+SO4•H2O, is a high spin
BASIC INFORMATION ON STRUCTURE AND BONDING compound with spin S = 2 and shows a large quadrupole splitting of ca.
3 mm s-1. B is a low spin compound with S = 0 and cubic (Oh) molecular
symmetry and shows no quadrupole splitting. C is also low spin with S = 0,
Quadrupole splitting in three typical iron(II) compounds: but strong tetragonal distortion from Oh symmetry due to the replacement of
one of the six CN- ligands by NO, which gives rise to a significant quadrupole
Figure 14 shows the Mössbauer spectra of three selected splitting. The occurrence of quadrupole splitting in A and C and the absence
iron(II) compounds: of it in B are explained in Figure 15 and Figure 16.
10UNTERRICHT
DEUTSCHE BUNSEN-GESELLSCHAFT
(A) FeSO4∙7H2O;
(B) K4[Fe(CN)6];
(C) Na2[Fe(CN)5NO].
Ferrous sulphate A, better formulated as [Fe(H2O)6]2+SO4∙H2O,
is a high spin compound with spin S = 2 and shows a large
quadrupole splitting of ca. 3 mm s-1. K4[Fe(CN)6] is a low spin
compound with S = 0 and cubic (Oh) molecular symmetry and
shows no quadrupole splitting. Na2[Fe(CN)5NO] is also low spin
with S = 0, but strong tetragonal distortion from Oh symmetry
due to the replacement of one of the six CN- ligands by NO
gives rise to a significant quadrupole splitting. The occurrence
of quadrupole splitting in A and C and the absence of it in B
are explained in Figure 15 and Figure 16. For high spin Fe2+
with 3d6 electron configuration, the six 3d electrons are distrib-
uted under Oh symmetry as shown in Figure 15 (left). The two
degenerate eg orbitals carry one electron each, and the three
degenerate t2g orbitals are occupied by 11/3 electrons on aver- Figure 16. K4[Fe(CN)6] is a 3d6 low spin complex with Oh symmetry, where
age. As the eg and t2g orbitals are cubic subgroups, there is no all six electrons are accommodated in the three t2g orbitals. Both contri-
butions (EFG)val and (EFG)lat vanish; there is no quadrupolar interaction.
valence electron contribution to the EFG independent of the Na2[Fe(CN)5NO] has C4v symmetry with d-orbital splitting as shown on the
number of electrons occupying them. There is also no lattice right. All six electrons are accommodated in the lowest three orbitals. (EFG)val
contribution to the EFG arising from the coordination sphere is still zero, but (EFG)lat ≠ 0 arises from the ligand replacement.
of six identical H2O ligands. Thus, there is no quadrupole split-
ting expected under Oh symmetry. [Fe(H2O)6]2+, however, is a plex with Oh symmetry, where all six electrons are accommo-
“Jahn-Teller-active” complex ion. It is unstable under Oh sym- dated in the three t2g orbitals. Both contributions (EFG)val and
metry. It undergoes axial distortion with symmetry lowering to (EFG)lat vanish; there is no quadrupolar interaction. Compound
D4h as schematised in Figure 15 (right), either compressing or C has C4v symmetry with d-orbital splitting as shown in Figure
elongating the octahedron in z-direction. Compression is pre- 16 (right). The low spin behaviour of C requires that all six elec-
ferred because in this case the ground state is an orbital sin- trons are accommodated in the lowest three orbitals arising
glet with the doubly occupied xy orbital being lowest in energy. from the tetragonal splitting of the former cubic t2g (Oh) sub-
Suppose all d-orbitals are singly occupied, as for instance in group. (EFG)val is still zero, but (EFG)lat ≠ 0 arises from the ligand
the case of [Fe(H2O)6]3+ (high spin), (EFG)val would be zero. But replacement.
the sixth electron placed in the lowest xy orbital in the case Structure of Fe3(CO)12: In 1965, Erickson and Fairhall sug-
of [Fe(H2O)6]2+ accumulates more charge in the xy plane than gested, on the basis of single crystal X-ray diffraction, three
along the z-axis and, thus, causes a large (EFG)val ≠ 0 and the possible molecular structures for Fe3(CO)12 [25]. In all cases
observed quadrupole splitting. the iron atoms form a triangle, but with different surroundings
The absence of quadrupole splitting in K4[Fe(CN)6] as well by the CO groups. In the upper two structures the three iron at-
as the relative large quadrupole splitting in Na2[Fe(CN)5NO] is oms have identical surroundings, the Mössbauer spectrum is
explained in Figure 16. K4[Fe(CN)6] (B) is a 3d6 low spin com- expected to show only one type of resonance signal. The lower
structure has two identical iron positions and a different one
for the third iron atom. In this case the Mössbauer spectrum
is expected to show two different types of resonance signals
with an area ratio of 2:1. A Mössbauer effect study performed
by Greatrex and Greenwood in 1969 [26] indeed showed two
types of resonance signals, a quadrupole doublet A and a sin-
glet B with an area ratio of 2:1 confirming the presence of two
types of iron positions in Fe3(CO)12 (Figure 17).
Figure 17. On the basis of single crystal X-ray diffraction, three possible
molecular structures for Fe3(CO)12 were suggested [25]. In the upper two
structures the three iron atoms have identical surroundings, the Mössbauer
Figure 15. Quadrupole splitting in the [Fe(H2O)6]2+complex ion arises from a spectrum is expected to show only one type of resonance signal. The lower
non-cubic valence electron distribution due to Jahn-Teller distortion with lo- structure has two identical iron positions and a different one for the third
wering of symmetry from Oh (EFG = 0) to D4h with valence electron population iron atom a different one for the third iron atom which confirmed by Möss-
in a compressed octahedron as shown in the figure and described in the text. bauer spectroscopy [26].
11UNTERRICHT
BUNSEN-MAGAZIN · 12. JAHRGANG · 1/2010
Effect of π-backdonation in [Fe(CN)5Xn-](3+n)-: The follow-
ing example demonstrates that Mössbauer spectroscopy can
help to characterise chemical bond properties. Taking from the
literature [11] the isomer shift data for the pentacyano com-
plexes of iron(II) with a different sixth ligand X and normalis-
ing the isomer shifts to that of the pentacyanonitrosylferrate
complex as zero point, one finds the ordering given in Table 2
which expresses the varying effects of dπ-pπ backdonation for
the different sixth ligand X.
Table 2. Effect of p-Backdonation in [Fe(CN)5Xn-](3+n)-
Figure 18. The graph shows the influence of electronegativity on the isomer
shift of ferrous halides. The electronegativity increases from iodine to flu-
orine. In the same ordering the 4s-electron population decreases and as a
direct consequence the s-electron density a the iron nucleus decreases, and
due to the fact that (Ra2 –Rg2) < 0 for 57Fe the isomer shift increases from
iodide to fluoride.
The isomer shift values become more positive on going
from NO+ to H2O. The reason is that in the same ordering the
strength of dπ-pπ backdonation decreases causing an increas-
ing d-electron density residing near the iron centre and thus
effecting stronger shielding of s-electrons by d-electrons, which
finally creates lower s-electron density at the nucleus in the
case of H2O as compared to NO+. The fact that the nuclear fac-
tor ΔR/R is negative for 57Fe explains the increasingly positive
isomer shift values in the given sequence from NO+ to H2O.
Effect of ligand electronegativity: In Figure 18 isomer
shift values of ferrous halides taken from the literature [11] Figure 19. In iron(II) compounds with relatively weak ligands coordinated
to the iron ions, e.g. water molecules, the six 3d electrons are accommo-
are plotted as a function of Pauling electronegativity values. dated spin-free according to Hund’s rule of maximum spin of S = 2. Such
The electronegativity increases from iodine to fluorine. In the compounds, called high spin complexes, are paramagnetic and are generally
same ordering the 4s-electron population decreases and as a weakly coloured. In Fe(II) compounds with relatively strong ligands like CN–
ions, the six electrons are arranged spin-paired with total spin S = 0. Such
direct consequence the s-electron density at the iron nucleus compounds are called low spin complexes; they are generally diamagnetic
decreases, and due to the fact that (Ra2 – Rg2) < 0 for 57Fe the and somewhat coloured. If the right kinds of ligands are chosen, e.g. deriva-
tives of tetrazole or triazole, one may observe spin state transition solely by
isomer shift increases from iodide to fluoride. varying the temperature, applying pressure or under irradiation with light as
shown in the cartoon. Increasing the temperature favours the HS state, ap-
plication of pressure favours the LS state. Green light (ca 500 nm) converts
LS to HS, red light (ca. 800 nm) HS to LS [27, 28].
SWITCHABLE MOLECULES: SPIN CROSSOVER
Thermally induced spin state transition from a high spin spin complexes, are paramagnetic and are generally weakly col-
(HS) state with maximum unpaired electrons to a low spin (LS) oured. In Fe(II) compounds with relatively strong ligands like
state with minimum unpaired electrons occurs in transition CN– ions, the six electrons are arranged spin-paired with total
metal compounds with d4 up to d7 electron configurations, if the spin S = 0. Such compounds are called low spin complexes;
difference between the energies of the HS and the LS states they are generally diamagnetic and often coloured. If the right
are comparable to thermal energy: ΔEHL ≈ kBT (kB = Boltzmann kinds of ligands are chosen, e.g. derivatives of tetrazole or tri-
constant). Figure 19 sketches on an elemental level the phe- azole, one may observe spin state transition solely by varying
nomenon in the case of iron(II) compounds with 6 electrons in the temperature, applying pressure or under irradiation with
the 3d valence shell. In iron(II) compounds with relatively weak light as shown in the cartoon.
ligands coordinated to the iron ions, e.g. water molecules, the Thermal spin crossover in iron(II) compounds is reflected by
3d electrons are accommodated spin-free according to Hund’s changes in the electron configuration. In the notation of ligand
rule of maximum spin of S = 2. Such compounds, called high field theoretical concepts, the electron configuration changes
12UNTERRICHT
DEUTSCHE BUNSEN-GESELLSCHAFT
from (t2g)4(eg)2 in the HS state to (t2g)6 in the LS state. This phase The influence of the ligand molecules on the spin state of
transition between paramagnetic and (nearly) diamagnetic is the central iron(II) ions is demonstrated with the two examples
easily detected by magnetic susceptibility measurements. As and their temperature dependent Mössbauer spectra shown in
the colour changes simultaneously, too, the transition from Figure 21. [FeII(phen)3]X2 (phen = 1.10-phenanthroline) is a typi-
one spin state to the other is also easily detected by optical cal low spin compound at all temperatures under study as con-
means. As indicated in Figure 19, the spin transition can also firmed by the characteristic Mössbauer spectra on the left with
be affected by applying external pressure or by irradiating the isomer shift of ca. 0.2 and quadrupole splitting ca. 0.5 mm s-1
material with light, where green light converts the LS to the HS independent of temperature. If one of the relatively strong phen
state and red light the HS to the LS state. This has become ligands, which occupies two coordination positions of the oc-
known as Light-Induced Excited Spin State Trapping (LIESST) tahedron, is replaced by two monofunctional NCS- groups, the
and reverse-LIESST [27, 28]. average ligand field strength becomes weaker than the mean
The condition that has to be met in order to observe thermal spin pairing energy and the compound [FeII(phen)2(NCS)2]
spin crossover is sketched in Figure 20 using the term symbols adopts high spin character at room temperature. The Mössbau-
known from ligand field theory. Thermal spin crossover may be er spectrum at 300 K shows the typical features of an iron(II)
observed if the ligand field strength of an iron(II) compound HS compound with isomer shift of ca. 1 mm s-1 and a large
is such that the difference between the lowest “vibronic” en- quadrupole splitting of ca. 3 mm s-1. However, the compound
ergy levels of the high spin state 5T2 and the low spin state 1A1 [FeII(phen)2(NCS)2] fulfils the condition for thermal spin crosso-
state is comparable with thermal energy kBT (kB = Boltzmann ver to occur, viz. ΔEHL » kBT. As the temperature is lowered, the
constant). The spin transition behaviour can be influenced by compound changes spin state from high spin to low spin near
chemical alteration of the material, e.g. ligand replacement, 180 K as is well documented by the Mössbauer spectra as a
change of non-coordinating anion and solvent molecule, sub- function of temperature on the right hand side of Figure 21,
stitution of spin state changing metal by another metal. For a which was first reported by Dezsi et al. in 1967 [29]. Since then
comprehensive coverage of chemical and physical influences more than 200 spin crossover compounds of iron(II) and iron(III)
on the spin transition behaviour see [27]. have been studied by Mössbauer spectroscopy (e.g. [28]).
[Fe(ptz)6](BF4)2, where ptz stands for the ligand molecule
1-propyl-tetrazole, is another iron(II) coordination compound ex-
hibiting thermal spin crossover with a spin transition tempera-
ture T1/2 of ca. 135 K. The 57Fe Mössbauer spectra clearly indi-
cate the transition at this temperature between the HS phase
(Figure 22; quadrupole doublet shown in red) and the LS phase
(singlet shown in blue). With this compound it was observed
for the first time that the spin transition can also be induced
by irradiating the crystals with light; green light converts the LS
state to the HS state, which can have very long lifetimes, e.g. on
the order of days at temperatures below ca. 20 K [30].
Figure 21. [FeII(phen)3]X2 (phen = 1.10-phenanthroline) is a typical low
spin compound with characteristic Mössbauer spectra shown on the left.
If one of the relatively strong phen ligands is replaced by two monofunc-
tional NCS- groups, the average ligand field strength becomes weaker than
Figure 20. Thermal spin crossover may be observed if the ligand field the mean spin pairing energy and the compound [FeII(phen)2(NCS)2] adopts
strength of an iron(II) compound is such that the difference between the high spin character at room temperature. The Mössbauer spectrum at
lowest “vibronic” energy levels of the high spin state 5T2 and the low spin 300 K on the right shows the typical features of an iron(II) HS compound.
state 1A1 state is comparable with thermal energy kBT (kB = Boltzmann con- [FeII(phen)2(NCS)2] fulfils the condition for thermal spin crossover to occur,
stant). Spin transition can also be induced by irradiation with light, applying viz. ΔEHL ≈ kBT. As the temperature is lowered, the compound changes from
pressure or a magnetic field. high spin to low spin near 180 K.
13Sie können auch lesen

















































