Erkrankungen durch genetische Mosaike - Übersichtsarbeit - Deutsches Ärzteblatt
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
MEDIZIN
Übersichtsarbeit
Erkrankungen durch genetische Mosaike
Ute Moog, Ute Felbor, Cristina Has, Birgit Zirn
B
ei genetisch bedingten Erkrankungen liegt die
Zusammenfassung krankheitsverursachende genetische Veränderung
meist in allen Körperzellen vor. Von einem Mosa-
Hintergrund: Genetische Mosaike entstehen durch Neumutationen, die sich erst
ikstatus spricht man hingegen, wenn nicht alle Körper-
nach der Befruchtung (postzygotisch) ereignen. Mosaike wurden in den letzten
zellen die gleiche genetische Ausstattung haben und in
Jahren als Ursache einer größeren Zahl von Erkrankungen beschrieben. Es handelt
einem Teil der Zellen eine pathogene Veränderung vor-
sich vor allem um neurokutane Erkrankungen und syndromale Entwicklungs-
kommt. Erkrankungen durch genetische Mosaike, wie
störungen mit charakteristischem Phänotyp. Teilweise besteht eine Tumordisposition.
zum Beispiel durch postzygotisch entstandene Chro-
Der vorliegende Artikel gibt einen Überblick über ausgewählte Mosaik-Erkrankungen.
mosomenfehlverteilungen, sind schon lange bekannt.
Methode: Es erfolgte eine selektive Literaturrecherche in PubMed mit besonderem Eine Mosaik-Trisomie 21 etwa wurde bereits 1961 be-
Fokus auf hochrangig publizierte, aktuelle Arbeiten zu den Themen asymmetrische schrieben (1). Prinzipiell kann auch jede monogene Er-
Wachstumsstörungen, fokale Hirnfehlbildungen, Mosaik-Erkrankungen aufgrund einer krankung als Mosaik vorliegen und dann mit einer mil-
Fehlregulation des RAS/RAF-Signalwegs (Mosaik-RASopathien) und vaskuläre deren oder atypischen Verlaufsform einhergehen als bei
Malformationen. Mutationen, die alle Körperzellen betreffen.
Neue Verfahren der Genomanalyse, besonders das
Ergebnisse: Die Identifizierung postzygotischer Mutationen hat zur Reklassifizierung
2005 eingeführte Verfahren der Hochdurchsatzsequen-
traditioneller Krankheitsbilder geführt und zu einem besseren pathogenetischen
zierung „next generation sequencing“ (NGS), haben
Verständnis beigetragen. Die Diagnosestellung wird durch den Einsatz moderner
den Nachweis von Mosaiken und hierdurch das Ver-
„next generation sequencing“(NGS)-Verfahren erleichtert, mit denen auch niedrig-
ständnis von Mosaik-Erkrankungen stark vorangetrie-
gradige Mosaike festgestellt werden können. Viele Mosaik-Mutationen sind nicht
ben (2). Pionierarbeit wurde bei Erkrankungen geleis-
im Blut nachweisbar, sondern nur im betroffenen Gewebe, zum Beispiel der Haut.
tet, die ausschließlich in Mosaikform vorzukommen
Genetische Mosaik-Erkrankungen manifestieren sich häufig an der Haut und am
scheinen. Sie werden durch Mutationen in zentralen
Gehirn sowie durch faziale Dysmorphien, asymmetrische Wachstumsstörungen und
Gefäßfehlbildungen.
Signalwegen hervorgerufen, die letal wären, wenn sie
konstitutionell (in allen Zellen) vorlägen (3, 4). Diese
Schlussfolgerung: Besonders bei asymmetrischen Wachstumsstörungen, Erkrankungen äußern sich klinisch häufig durch einen
fokalen neuronalen Migrationsstörungen, Gefäßfehlbildungen und linienförmigen partiellen Großwuchs, fokale Hirnfehlbildungen, kuta-
Hauterscheinungen sollte bei der Diagnostik an eine Mosaik-Erkrankung gedacht ne Symptome und Gefäßfehlbildungen. Jedes Krank-
werden. Der Nachweis eines postzygotischen Mosaiks bedeutet für Eltern eines heitsbild für sich ist selten, zusammen bilden sie aber
betroffenen Kindes oft eine Entlastung, da für weitere Kinder in der Regel kein eine wachsende Gruppe von erkennbaren Krankheits-
erhöhtes Wiederholungsrisiko besteht. Die korrekte Klassifizierung ist wichtig, da bildern mit richtungsweisenden Auffälligkeiten (5–8).
für einige Mosaik-Erkrankungen, zum Beispiel für „PIK3CA-related overgrowth Nicht selten besteht eine Tumordisposition.
spectrum“ (PROS) mit einem PIK3CA-Inhibitor bereits molekulare Therapieansätze Der Nachweis einer Mosaik-Erkrankung kann thera-
zur Verfügung stehen. peutisch relevant sein, erfordert aber einen gerichteten
Verdacht und eine geeignete Methodenwahl bei der ge-
Zitierweise
netischen Diagnostik, da der Nachweis meist nicht im
Moog U, Felbor U, Has C, Zirn B: Disorders caused by genetic mosaicism.
Dtsch Arztebl Int 2020; 117: 119–25. DOI: 10.3238/arztebl.2020.0119
Blut gelingt. Dieser Artikel gibt, ohne Anspruch auf
Vollständigkeit zu erheben, einen Überblick über Krank-
heiten durch genetische Mosaike und möchte aufzeigen,
wann eine Mosaik-Erkrankung in Betracht gezogen wer-
den sollte. Er stellt ausgewählte Krankheitsbilder vor
und erläutert das diagnostische Prozedere.
Methode
Die Darstellung der Krankheitsgruppen und die Aus-
wahl der beschriebenen Entitäten beruht auf unserer
Institut für Humangenetik, Universitätsklinikum Heidelberg: Prof. Dr. Dr. med. Ute Moog Expertenmeinung. Für folgende Begriffe führten wir
Institut für Humangenetik, Universitätsmedizin Greifswald und Interfakultäres Institut für Genetik und
eine selektive Literaturrecherche in PubMed durch,
Funktionelle Genomforschung, Universität Greifswald: Prof. Dr. med. Ute Felbor jeweils unter gleichzeitiger Verwendung des Begriffs
Klinik für Dermatologie und Venerologie, Universitätsklinikum Freiburg, Medizinische Fakultät, „genetic“: „focal cortical dysplasia“ (90 Treffer),
Albert-Ludwigs-Universität Freiburg: Prof. Dr. med. Cristina Has „hemimegalencephaly“ (29 Treffer), „mosaic RASopathy“
®
genetikum , Genetische Beratung und Diagnostik, Stuttgart: Prof. Dr. med. Dr. rer. nat. Birgit Zirn (4 Treffer), „PIK3CA related overgrowth spectrum“
Deutsches Ärzteblatt | Jg. 117 | Heft 8 | 21. Februar 2020 119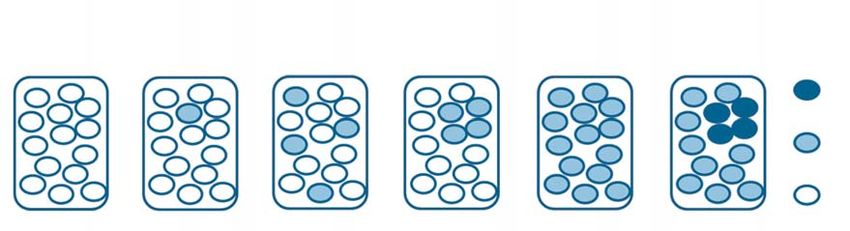
MEDIZIN
GRAFIK 1
normales Allel in
homozygotem Zustand
mutiertes Allel in
heterozygotem Zustand
zwei mutierte Allele
gesund punktuelles disseminiertes segmentales autosomal segmentales
Mosaik Mosaik Mosaik Typ 1 dominante Mosaik Typ II
Erkrankung
Schematische Darstellung von Mosaiktypen. Jedes Rechteck stellt ein Individuum dar. Die Ellipsen repräsentieren einzelne Zellen.
Weiß steht für normale Allele. Hellblau steht für Heterozygotie für ein mutiertes Allel; dunkelblau repräsentiert das Auftreten eines zweiten Mutationsereignisses
bei einem Individuum mit einer heterozygoten Mutation und einer autosomal dominanten Erkrankung (modifiziert nach [7]).
(10 Treffer), jeweils AND „review“; „port-wine stain“ ● Mosaike für Mutationen, die bei autosomal domi-
AND „Sturge Weber syndrome“ (7 Treffer), „capillary nanten Erkrankungen bekannt sind. Diese Mosaike
malformation-arteriovenous malformation (CM-AVM)“ treten abhängig vom Zeitpunkt des Mutationser-
AND „vascular“ (43 Treffer), jeweils AND „mutation“. eignisses entweder disseminiert auf (Grafik 1) und
Nach der Korrektur von Redundanzen wurden insge- führen dann zu atypischen oder attenuierten For-
samt 184 Literaturstellen berücksichtigt. men eines Krankheitsbildes, oder lokalisiert als
segmentales Mosaik Typ I (Grafik 1) mit meist
Genetische Mosaike milderen Auswirkungen (4). Beispiele sind die
Mosaike entstehen durch spontane Neumutationen segmentale Neurofibromatose Typ 1 (NF1) oder
meist in der frühen embryonalen oder fetalen Entwick- Mosaikformen der tuberösen Sklerose (13, 14).
lung (9). Somit handelt es sich nicht um vererbte Muta- ● Seltene Mosaike, die bei autosomal dominant erb-
tionen, die schon in der Eizelle oder im Spermium vor- lichen Erkrankungen durch ein zweites Mutations-
liegen, sondern um postzygotische, das heißt nach der ereignis auf dem anderen Allel (meistens Verlust
Befruchtung auftretende Ereignisse. Für Eltern eines der Heterozygotie) zu einer Aggravation des Phä-
betroffenen Kindes ist die Information über das Vorlie- notyps in einem segmentalen Bereich führen (seg-
gen einer postzygotischen genetischen Veränderung mentales Mosaik Typ II) (Grafik 1) (4, 12).
wichtig, da in diesem Fall kein erhöhtes Wiederho- Hinweise auf Mosaik-Erkrankungen können sichtba-
lungsrisiko für dieselbe Erkrankung bei weiteren Kin- re, persistierende Veränderungen der Haut sein, die
dern besteht. Das Kind kann die Mutation seinerseits punktuell, disseminiert, segmental oder linear vorlie-
nur an die nächste Generation vererben, wenn auch sei- gen. Das häufigste Verteilungsmuster postzygotischer
ne Keimzellen (Ei- oder Samenzellen) von dem Mosaik Mosaike sind die Blaschko-Linien, ein Liniensystem
betroffen sind. Im Vererbungsfall tritt dann allerdings der Haut, das der Migration der Zellen während der
bei Nachkommen kein Mosaik, sondern eine durchgän- Embryogenese entspricht (e1, e2). Beispielsweise kön-
gige Mutation auf. nen Pigmentmosaike bei Chromosomenstörungen so-
Der Schweregrad und die klinische Symptomatik wie isolierte oder syndromale epidermale Nävi (Abbil-
postzygotischer Mosaike sind vom Zeitpunkt des Mu- dung 1) den Blaschko-Linien folgen.
tationsereignisses, der Zellart, in der die Mutation statt- Die Haut ist wahrscheinlich deshalb ein häufiges
findet, der Expansion von Zellen mit Mutation, dem Manifestationsorgan von Mosaiken, weil kutane Verän-
mutierten Gen und der Mutation abhängig (3). Je später derungen leicht zugänglich und dann diagnostisch ver-
Mosaike in der embryonalen Entwicklung auftreten, wertbar sind. Auch verschiedene der im Folgenden auf-
umso begrenzter ist die Symptomatik. Beispielsweise geführten Mosaik-Erkrankungen gehen mit Hautverän-
gehen bestimmte Nävi auf lokale Mosaike in Hautzel- derungen einher.
len zurück (10, 11).
Mosaike können folgendermaßen eingeteilt werden: Asymmetrische Wachstumsstörungen
● Mosaike für letale Mutationen führen zu Krank- Asymmetrische Wachstumsstörungen können durch
heitsbildern, die nur in Mosaikform existieren, aktivierende („gain of function“-)Mosaik-Mutationen
wie zum Beispiel das Proteus-, das Sturge-Weber- in Genen bedingt sein, die zu vermehrter Zellteilung
oder das McCune-Albright-Syndrom (12). Diese und somit zu verstärktem Gewebewachstum führen
Erkrankungen können somit auch nicht von den und im sogenannten PI3K/AKT-Signalweg zusammen-
Betroffenen an eigene Kinder vererbt werden, da wirken (Grafik 2; 7, 15). Vor allem das PIK3CA-Gen
im Vererbungsfall die Mutation durchgängig vor- (in der Signalkaskade ganz oben) ist häufig von
läge und letal wäre. Mosaik-Mutationen betroffen und führt zu einer äußerst
120 Deutsches Ärzteblatt | Jg. 117 | Heft 8 | 21. Februar 2020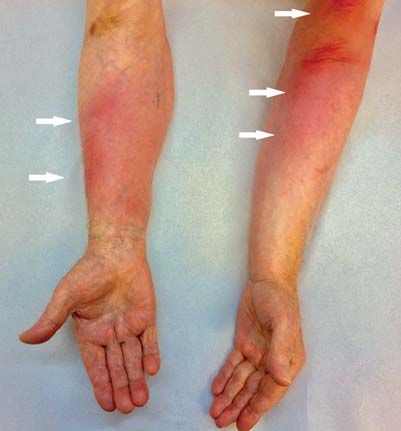
MEDIZIN
variablen phänotypischen Ausprägung, je nachdem wel- Abbildung 1: Mosaik-RASopathie durch
che Zellen und Gewebe betroffen sind. KRAS-Mutation im Mosaik bei einer
PIK3CA-Mutationen wurden 2012 als ursächlich für 21-jährigen Frau mit linearen Hyperpigmen-
tierungen und Nävi sebacei überwiegend
verschiedene, zuvor klinisch definierte Phänotypen be-
der linken Körperhälfte. Zudem bestanden
schrieben (16). Als Oberbegriff hat sich „PIK3CA-rela- ein schmächtigeres linkes Bein, eine
ted overgrowth spectrum“ (PROS) durchgesetzt (6, 17). Skoliose, ein haarloser Fettgewebsnävus
Zu dieser Entität gehört das „megalencephaly-capillary der Kopfhaut (sog. Nävus psiloliparus) und
malformation-polymicrogyria“(MCAP)-Syndrom (Ab- eine fibröse Dysplasie des linken Femurs
bildung 2), bei dem die asymmetrische Wachstumsstö- (nicht abgebildet). Die Mutation war an DNA
rung meist ab Geburt vorhanden ist und im Verlauf der aus betroffenem Gewebe der Kopfhaut, nicht
Kindheit weiter zunimmt. Betroffene Kinder weisen aber an Blut-DNA nachweisbar.
häufig eine Makrozephalie und kapilläre Gefäßfehl-
bildungen (Nävus flammeus) sowie eine marmorierte
Haut auf. Zudem können eine Hemimegalenzephalie,
ein asymmetrischer Hydrozephalus und variable Gyrie-
rungsstörungen, vor allem eine Polymikrogyrie, vor-
handen sein. Hierdurch erklärt sich auch das Risiko für
eine meist schwierig zu therapierende Epilepsie und ei-
ne Entwicklungsstörung beziehungsweise Intelligenz-
minderung. oder zu einer fokal vorliegenden neuronalen Migrations-
Der Nachweis von PIK3CA-Mutationen im Mosaik störung, einer fokalen kortikalen Dysplasie (FCD) (19).
ist meist nicht im Blut, sondern nur an DNA aus betrof- Diese beiden Störungen gehören zu einem zerebralen
fenem Gewebe möglich. Besonders gut sind Hautbiop- Fehlbildungsspektrum, dessen Manifestationen vom
sien geeignet. Nur bei circa 10 % kommen konstitutio- Zeitpunkt des postzygotischen Auftretens der Mutation
nelle PIK3CA-Mutationen vor, die alle Zellen betreffen und von der betroffenen neuronalen Vorläuferzelle
(Nicht-Mosaik). In diesen Fällen prägt sich die Wachs- abhängig sind (8, 20). Häufig ist auch eine therapie-
tumsstörung nicht asymmetrisch aus (e3). Wahrschein- refraktäre fokale Epilepsie im Kindesalter mit ihnen
lich haben die meisten konstitutionellen PIK3CA-Mu- verbunden. Postzygotische Mutationen werden bei bis
tationen eine sehr schwere Ausprägung und sind letal. zu 30 % der Patienten mit neuronalen Migrations-
Das Proteus-Syndrom ist ein Beispiel für eine asym- störungen vermutet (8, e4).
metrische Wachstumsstörung, die sich erst im Laufe FCD oder HME sind wichtige Indikationen für epi-
der Kindheit zeigt. Im Alter von 2–4 Jahren beginnen lepsiechirurgische Eingriffe bei Kindern (21). Geneti-
einzelne Körperregionen, zum Beispiel Zehen oder ei- sche Studien an betroffenem Hirngewebe haben bei
ne ganze Extremität, übermäßig zu wachsen. Vor allem FCD Typ2/HME aktivierende somatische Mutationen
an den Fußsohlen kann ein Nävus cerebriformis mit in MTOR und seinen Aktivatoren (Grafik 2, zum Bei-
überschießendem Gewebewachstum, das oberflächlich spiel PIK3CA) nachgewiesen sowie LOF-Mutationen
ein Bild wie Hirnfurchungen ergibt, auftreten. Im Ver- in verschiedenen negativen Regulatoren des Signal-
lauf kommt es häufig zu schwersten Beeinträchtigun- wegs, die auch als Keimbahnmutationen vorliegen kön-
gen durch das mosaikartig vermehrte Wachstum am ge- nen (8, 19, 21). Bei FCD Typ1 hingegen liegen in circa
samten Körper. Die Intelligenz ist typischerweise nor- 30 % der Fälle postzygotische Varianten in einem Gly-
mal. Das Proteus-Syndrom ist durch eine spezifische kosylierungs-Gen (SLC35A2) vor (21). Die Mosaik-
Mosaik-Mutation im AKT1-Gen bedingt (18). Mutationen lassen sich in aller Regel nur in betroffe-
Der PI3K/AKT-Signalweg umfasst auch Gene, in nem Gewebe nachweisen, bei FCD oft niedriggradig,
denen „loss of function“(LOF)-Mutationen (Funktions- bei HME in bis zu 30 % (8, 21).
verlust mit Ausfall der „Bremse“) zu einem vermehrten
Zellwachstum führen (zum Beispiel PTEN, TSC1/2, Mosaik-RASopathien
Grafik 2). LOF-Mutationen in diesen Genen liegen Mit „RASopathien“ wird eine Gruppe von Krankhei-
häufiger auf einem Allel konstitutionell als erbliche ten um das Noonan-Syndrom bezeichnet, die auf ei-
Keimbahnmutation vor. Krankheitsspezifische Läsio- ner Fehlregulation des RAS/RAF-Signalwegs durch
nen entstehen erst, wenn das zweite Allel durch eine meist aktivierende, konstitutionelle Mutationen betei-
somatische, lokal begrenzte Mutation seine Funktion ligter Gene beruhen. Gemeinsame klinische Merkmale
verliert (Knudson’s Zwei-Treffer-Modell). sind:
● kardiovaskuläre Anomalien
Fokale Hirnfehlbildungen ● vermindertes Wachstum
Postzygotische Mutationen in Genen des PI3K/AKT- ● Dysmorphien
Signalwegs können nicht nur im Rahmen eines MCAP- ● Auffälligkeiten der Haut
Syndroms mit Hirnfehlbildungen einhergehen, sondern ● Entwicklungsstörungen in variabler Ausprägung.
auch isoliert oder im Rahmen anderer Krankheitsbilder Teils besteht auch eine Tumordisposition (22). Wie
zu asymmetrischem „Großwuchs“ von Hirngewebe füh- der PI3K/AKT-Signalweg, mit dem er vernetzt ist,
ren, besonders zu einer Hemimegalenzephalie (HME), stimuliert der RAS/RAF-Signalweg das Zellwachstum
Deutsches Ärzteblatt | Jg. 117 | Heft 8 | 21. Februar 2020 121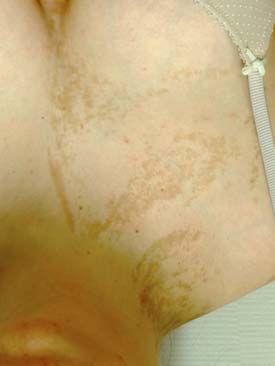
MEDIZIN
GRAFIK 2 letal wären, sollten sie konstitutionell bedingt sein
(5, 22). Eine Ausnahme sind HRAS-Mutationen.
S Der
Wachstumsfaktor
Mutationsnachweis gelingt meist nur an betroffenem
Gewebe. Die neurokutane Melanose betrifft einen
kleinen Teil der Patienten mit den häufigen kongeni-
Rezeptor talen melanozytären Nävi (CMN) und zeichnet sich
durch große (Riesen)-CMN in Kombination mit einer
PI3K/AKT-Signalweg RAS/RAF-Signalweg
oft symptomatischen leptomeningealen Melanozyto-
se aus (24). Sie wird durch eine frühembryonale
NRAS-Mutation
S im Neuroektoderm hervorgerufen.
PTEN PI3K RAS NF1
Das Risiko für Melanome ist von der Ausdehnung der
crosstalk CMN abhängig und beläuft sich auf circa 1 % bei
CMN allgemein gegenüber 12 % bei Riesen-CMN
AKT RAF (25). Bei der neurokutanen Melanose besteht auch ein
erhöhtes Risiko für ZNS-Melanome. Lokal begrenzte
TSC mTOR MEK
postzygotische Mutationen in unterschiedlichen Ge-
nen führen zu weiteren Nävi, zum Beispiel einem ke-
ratinozytischen Epidermalnävus oder einem Nävus
ERK sebaceus.
Treten die Mutationen früher in der Entwicklung
auf und betreffen mehrere Gewebe, kommt es zu syn-
Zellteilung/Wachstum ↑ Regulation
Angiogenese ↑ Zellzyklus/Apoptose dromalen Krankheitsbildern mit dem Leitsymptom
eines bestimmten Nävus (3). Zu nennen ist hier das
klinisch seit Jahrzehnten bekannte Schimmelpenning-
PI3K/AKT- und RAS/RAF-Signalweg mit beteiligten Hauptgenen. Feuerstein-Mims-Syndrom (SFMS), bei dem beson-
Nach der Bindung eines Wachstumsfaktors an den zugehörigen Rezeptor, zum Beispiel Re-
zeptor Tyrosinkinase (RTK), „Insulin growth factor 1 Rezeptor“ (IGF-1R), werden beide Signal- ders das ZNS, die Augen und das Skelett (Osteomala-
wege aktiviert. Zudem besteht eine Wechselwirkung („crosstalk“) zwischen mehreren nachfol- zie, hypophosphatämische Rachitis) betroffen sein
gend aktivierten Genen. können (26), mit Nachweis von Mosaik-Mutationen
Der PI3K/AKT-Signalweg (links) führt zu vermehrter Zellteilung und Angiogenese. Das Gen
PIK3CA A kodiert für eine katalytische Untereinheit (p110α) der Phosphatidylinositol-3-Kinase
in HRAS, KRAS S und NRAS S (10). Die beachtliche Va-
(PI3K). Mutationen in PIK3CA (meist im Mosaik vorliegend) führen zum sogenannten PIK3CA- riabilität der klinischen Symptomatik konnte durch
related overgrowth spectrum (PROS). Dazu gehören „megalencephaly capillary malformation” die Identifizierung als Mosaik-Erkrankung erklärt
((MCAP), ) Hemimegalenzephalie,
g p „fibroadipose
p overgrowth”
g (FAO)
( ) und „congenital
g lipomatous
p
overgrowth, vascular malformations, epidermal nevi, skeletal anomalies” (CLOVES). Über die
werden. Auch Syndrome mit angeborenen Anomalien
Proteinkinase AKT wird „mammalian target of rapamycin“ (mTOR) aktiviert, was zu vermehrter von Auge und Haut wurden als Mosaik-RASopathie
Zellteilung und Gewebewachstum führt. Mosaik-Mutationen im AKT1-Gen sind Ursache des identifiziert: das okulo-ektodermale Syndrom (OES,
Proteus-Syndroms und MTOR-Mutationen bedingen das Smith-Kingsmore-Syndrom sowie die Mutationen in KRAS), S dessen Hauptmerkmale Epi-
Hemimegalenzephalie und die fokale kortikale Dysplasie. Mutationen in den genannten Genen
sind „gain of function“-Mutationen. Antagonisten der Wachstumsaktivierung über die bulbärdermoide und angeborene Skalpdefekte sind
PI3K/AKT-Signalkaskade sind PTEN N (Cowden-Syndrom, Makrozephalie-Autismus-Syndrom, (27, 28), und die enzephalo-kranio-kutane Lipomato-
Bannayan-Riley-Ruvalcaba-Syndrom) und die tuberöse Sklerose (TSC1/2), deren Ausfall se (ECCL, Mutationen in FGFR1 und KRAS), S bei
durch „loss of function“-Mutationen ebenfalls zu einer Wachstumssteigerung führt. Diese Muta-
tionen liegen meist nicht im Mosaik, sondern konstitutionell vor. der zusätzlich im ZNS die namengebenden Lipome
Der RAS/RAF-Signalweg (rechts) reguliert den Zellzyklus, die Zelldifferenzierung und die vorliegen (29, 30). Beide Syndrome können weitere
Apoptose. Mutationen in beteiligten Genen führen zu den sogenannten RASopathien (Noo- Auffälligkeiten der Haut, unter anderem einen Nävus
nan-Syndrom: Gene PTPN11, SOS1, RAF1, KRAS, NRAS, SHOC2, CBL; Costello-Syndrom:
HRAS-Gen; „cardio-facio-cutanes“(CFC)-Syndrom: BRAF, MAP2K1/2). / In den genannten Ge- sebaceus, aufweisen. Weiterhin umfassen sie eine Dis-
nen finden sich „gain
g of function“-Mutationen. Neurofibromatose Typ yp 1 ((NF1)) ist ein negativer
g position für Kiefertumore, nicht ossifizierende Fibrome
Regulator des RAS/RAF-Signalwegs. Die phänotypische Überlappung ist wie bei den Syndro- und, im Falle einer ECCL, auch für niedriggradige
men des PI3K/AKT-Signalweges sehr groß (7, 15).
Gliome (31). Da es nicht immer möglich ist, bisher ver-
wendete klinische Diagnosen eindeutig zuzuordnen,
wird die Bezeichnung (KRAS
( S-assoziierte) Mosaik-RA-
und wird nach Bindung von Wachstumsfaktoren an Sopathie oft zutreffender sein (Abbildung 1).
Rezeptoren auf der Zelloberfläche durch RAS-Protei-
ne vermittelt (Grafik 2). Die klassischen RAS-Protei- Gefäßmalformationen
ne werden von den Genen HRAS, KRAS S und NRAS Die diagnostische Abgrenzung des isolierten Portwein-
kodiert. Somatische Mutationen in diesen Genen, die flecks vom sporadisch auftretenden Sturge-Weber-
spontan in einzelnen Zellen auftreten und aufgrund Syndrom (SWS) mit fazialem Nävus flammeus, intra-
eines Wachstumsvorteils zu einer klonalen Expansion kraniellen und intraokulären Gefäßfehlbildungen ist
führen, spielen in der Onkogenese eine weitverbreite- prognostisch bedeutsam. Beim SWS kann die leptome-
te Rolle (23). ningeale Angiomatose bereits im ersten Lebensjahr zu
Einen anderen Phänotyp als die konstitutionellen epileptischen Krisen führen, die die psychomotorische
RASopathien haben die Mosaik-RASopathien, die und geistige Entwicklung beeinflussen. Schlaganfall-
auf postzygotischen „gain-of-function“-Mutationen artige Episoden und das Glaukom gehören ebenfalls zu
in RAS/RAF-Genen
F beruhen, die anzunehmenderweise den schwerwiegenden Manifestationen.
122 Deutsches Ärzteblatt | Jg. 117 | Heft 8 | 21. Februar 2020MEDIZIN
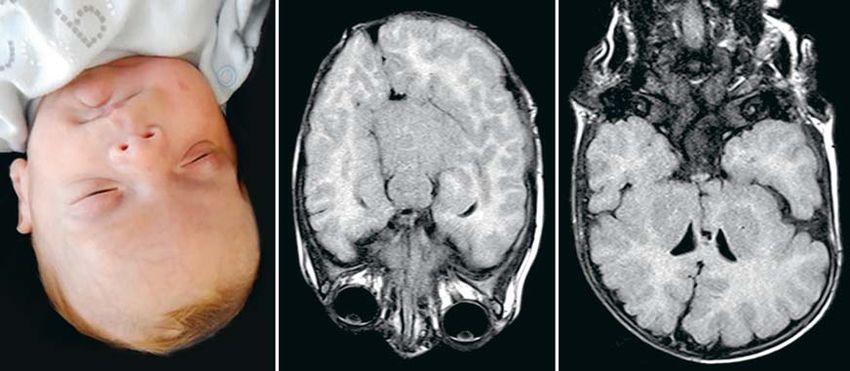
Abbildung 2:
MCAP-Syndrom
durch PIK3CA-Mutation
im Mosaik bei einem
5 Monate alten
Jungen. Fazialer
Phänotyp mit
deutlicher Asymmetrie
und vermehrtem
Wachstum der linken
Wange sowie cMRT
mit deutlicher Hemi-
megalenzephalie links.
MCAP, „megalence-
phaly-capillary malfor-
mation-polymicrogy-
ria“; MRT, Magnet-
resonanztomografie
Ätiologisch sind der häufige Portweinfleck und das Analyse mittels NGS kann prinzipiell an DNA unter-
seltene SWS zwei extreme klinische Ausprägungen schiedlicher Herkunft (zum Beispiel Blut, Fibroblasten,
desselben molekularen Mechanismus. In fehlgebilde- Urinsediment, Hirngewebe) durchgeführt werden. Be-
ten Kapillaren wird bei beiden Krankheitsbildern in sonders gut eignet sich DNA aus Fibroblasten. Eine
der Mehrzahl der Fälle eine spezifische postzygoti- Hautbiopsie kann beispielsweise im Rahmen anderer
sche Punktmutation im GNAQ-Gen identifiziert, die geplanter Eingriffe entnommen werden. Bezüglich
den RAS/RAF-Signalweg aktiviert (32). Wahrschein- weiterer Spezialverfahren verweisen wir auf eine 2018
lich entstehen nichtsyndromale Portweinflecken publizierte Übersichtsarbeit (8).
durch eine späte und das SWS durch eine frühe post-
zygotische Mutation. Bei sporadischen vaskulären Therapie
Malformationen wurden ebenfalls Mutationen in RAS Der PI3K/AKT-Signalweg ist bereits Ansatz verschie-
und anderen Genen des RAS/RAF-Signalwegs identi- dener Therapiestrategien und zahlreiche Medikamente
fiziert (33). sind in Entwicklung. Vor allem wird dabei auf eine
Roséfarbene kapilläre Malformationen der Haut Hemmung von mTOR gezielt, das die Wachstumsstei-
in Kombination mit arteriovenösen Malformationen gerung von PIK3CA und AKT vermittelt (zum Beispiel
bei zumindest einem weiteren Familienangehörigen Everolimus/Sirolimus) (20, 36). Vielversprechende Er-
sowie gelegentlich assoziierten Hemihyperplasien bil- gebnisse für die Wachstumshemmung bei Patienten mit
den ein klinisch variables Krankheitsbild (CM-AVM), PROS zeigte kürzlich eine Studie zum oralen PIK3CA-
welches erst nach einer Identifizierung von LOF- Inhibitor BYL719, der zu einer signifikanten Volumen-
Varianten im RASA1-Gen beschrieben wurde (34). reduktion von läsionalem Gewebe (bei radiologischer
RASA1 ist ein Negativregulator von RAS. Folglich Kontrolle um 27,2 ± 14,6 beziehungsweise 37,8 ± 16,3 %
aktiviert ein Funktionsverlust ebenfalls den RAS/ nach drei beziehungsweise sechs Monaten) und einer
RAF-Signalweg. deutlichen Verbesserung der Lebensqualität führte (37).
Während das auf einer aktivierenden Punktmutati- Auch bei FCD/HME umfassen therapeutische Ansätze
on im GNAQ-Gen basierende SWS nur in Mosaikform mTOR- und PIK3CA-Inhibitoren sowie in SLC35A2-
beobachtet wird, können RASA1-assoziierte Phäno- assoziierten Fällen diätetische Maßnahmen (21). Bei
typen einem autosomal-dominanten Erbgang folgen. einer Patientin mit Proteus-Syndrom und Ovarialkarzi-
Zusätzlich zu der dominant vererbten Mutation in einer nom wurde über die Behandlung mit einem AKT-Inhi-
Genkopie führt eine zweite neu auftretende Mutation bitor berichtet (38).
der zweiten Genkopie zum Funktionsverlust des RASA1- Für Mosaik-Krankheiten des RAS/RAF-Signalwegs
Gens. werden bisher in Einzelfällen besonders MEK-Inhibi-
toren erprobt (25), bei großen CMN in präklinischer
Molekulargenetische Diagnostik Phase auch eine Kombination von MEK- und AKT-In-
Hochgradige genetische Mosaike können sich bei der hibitoren (39). Die Notwendigkeit einer ätiologischen
klassischen Sanger-Sequenzierung zeigen, indem eine Einordnung erscheint bei den Mosaik-RASopathien be-
zweite Base als zusätzlicher kleiner „peak“ im Chroma- sonders dringlich, da sich diese oft durch eine Tumor-
togramm erscheint. Deutlich zuverlässiger lassen sich disposition auszeichnen. Bei den sporadischen Gefäß-
Mosaike mittels NGS diagnostizieren und quantifizie- malformationen, die zu Schlaganfällen, Blutungen und
ren. Mit hoher Sequenziertiefe können hierbei auch anderen Komplikationen führen können, werden im
niedriggradige Mosaike festgestellt werden (35). Eine Tiermodell BRAF-Inhibitoren erforscht (33).
Deutsches Ärzteblatt | Jg. 117 | Heft 8 | 21. Februar 2020 123MEDIZIN
3. Lim YH, Moscato Z, Choate KA: Mosaicism in cutaneous disorders.
Annu Rev Genet 2017; 51: 123–41.
Kernaussagen 4. Happle R: The categories of cutaneous mosaicism: a proposed clas-
sification. Am J Med Genet A 2016; 170A: 452–9.
● Klinisch wegweisend für genetische Mosaike sind asymmetri-
5. Hafner C, Groesser L: Mosaic RASopathies. Cell Cycle 2013; 12:
sche Wachstumsstörungen in Kombination mit lokalisierten 43–50.
persistierenden Hautveränderungen (zum Beispiel Pigment- 6. Keppler-Noreuil KM, Rios JJ, Parker VE, et al.: PIK3CA-related over-
flecken, Nävi) besonders entlang der Blaschko-Linien und growth spectrum (PROS): diagnostic and testing eligibility criteria,
differential diagnosis, and evaluation. Am J Med Genet A 2015;
Gefäßfehlbildungen. 167A: 287–95.
● Die nosologische Abgrenzung von Mosaik-Erkrankungen ist 7. Nathan N, Keppler-Noreuil KM, Biesecker LG, Moss J, Darling TN:
Mosaic disorders of the PI3K/PTEN/AKT/TSC/mTORC1 signaling
schwierig. Sie zeichnen sich durch ein breites phänotypi- pathway. Dermatol Clin 2017; 35: 51–60.
sches Spektrum und große klinische Variabilität aus. 8. D‘Gama AM, Walsh CA: Somatic mosaicism and neurodevelop-
mental disease. Nat Neurosci 2018; 21: 1504–14.
● Mosaik-Erkrankungen entstehen als Neumutation postzygo- 9. Bae T, Tomasini L, Mariani J, et al.: Different mutational rates and
tisch bei den Betroffenen selbst und sind nicht vererbt. Sie mechanisms in human cells at pregastrulation and neurogenesis.
können von Betroffenen an eigene Kinder nur als durchgän- Science 2018; 359: 550–5.
10. Groesser L, Herschberger E, Ruetten A, et al.: Postzygotic HRAS
gige Mutation vererbt werden, wenn es sich um nicht-letale and KRAS mutations cause nevus sebaceous and Schimmelpenning
Mutationen handelt, die auch die Keimbahn betreffen. syndrome. Nat Genet 2012; 44: 783–7.
11. Happle R: Wie häufig sind genetische Mosaike in der Haut? Hautarzt
● Mosaike sind oft nicht im Blut, sondern nur in betroffenem 2014; 65: 536–41.
Gewebe nachweisbar. 12. Has C, König A: Rudolf Happle zum 80. Geburtstag. Hautarzt 2018;
69: 343–6.
● Therapeutische Ansätze stehen besonders für Gene des 13. Calì F, Chiavetta V, Ruggeri G, et al.: Mutation spectrum of NF1 gene
PI3K/AKT- und RAS/RAF-Signalwegs zur Verfügung. in Italian patients with neurofibromatosis type 1 using Ion Torrent
PGM™ platform. Eur J Med Genet 2017; 60: 93–9.
14. Peron A, Au KS, Northrup H: Genetics, genomics, and genotype-
phenotype correlations of TSC: insights for clinical practice. Am J
Med Genet C Semin Med Genet 2018; 178: 281–90.
15. Borrie SC, Brems H, Legius E, Bagni C: Cognitive dysfunctions in
Ausblick intellectual disabilities: the contributions of the Ras-MAPK and
Postzygotische Mutationen wurden besonders bei der PI3K-AKT-mTOR pathways. Annu Rev Genomics Hum Genet 2017;
18: 115–42.
erneuten Analyse von Exom-Daten auch bei 3–9 % von 16. Rivière JB, Mirzaa GM, O‘Roak BJ, et al.: De novo germline and
Patienten mit Entwicklungsstörungen ohne strukturelle postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spec-
zerebrale Auffälligkeiten nachgewiesen, zum Beispiel trum of related megalencephaly syndromes. Nat Genet 2012; 44:
934–40.
bei Autismus oder genetisch bedingter Epilepsie (8, 17. Mirzaa G, Timms AE, Conti V, et al.: PIK3CA-associated develop-
40). Methodisch bedingt, zum Beispiel durch ungenü- mental disorders exhibit distinct classes of mutations with variable
gende Sequenziertiefe, dürften aktuell noch viele auf expression and tissue distribution. JCI Insight 2016; 1: pii: 87623.
Mosaiken beruhende Erkrankungen dem Nachweis ent- 18. Lindhurst MJ, Sapp JC, Teer JK, et al.: A mosaic activating mutation
in AKT1 associated with the Proteus syndrome. N Engl J Med 2011;
gehen. Zudem kann die Zugänglichkeit von betroffe- 365: 611–9.
nem Gewebe die molekulare Analyse einer Mosaik-Er- 19. Juric-Sekhar G, Hevner RF: Malformations of cerebral cortex devel-
krankung limitieren. Bei fokalen Hirnfehlbildungen opment: molecules and mechanisms. Annu Rev Pathol 2019; 14:
293–318.
lässt sich der Nachweis einer Mosaik-Erkrankung all- 20. Marsan E, Baulac S: Review: Mechanistic target of rapamycin
gemein nur dann erbringen, wenn betroffenes Gewebe (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol
bei einer Operation gewonnen werden kann. Appl Neurobiol 2018; 44: 6–17.
Bei vielen Krankheitsbildern kommt es derzeit zu ei- 21. Baldassari S, Ribierre T, Marsan E, et al.: Dissecting the genetic basis
of focal cortical dysplasia: a large cohort study. Acta Neuropathol
ner molekularen Reklassifizierung. Dies ist nicht nur 2019; 138: 885–900.
für das Verständnis, sondern auch für einen eventuellen 22. Zenker M, Kutsche K: RASopathien. medgen 2016; 28: 15.
Einschluss in Studien und für die Therapie bedeutsam. 23. Li S, Balmain A, Counter CM: A model for RAS mutation patterns in
cancers: finding the sweet spot. Nat Rev Cancer 2018; 18: 767–77.
Die diagnostische Unterscheidung von Mosaik- und
24. Alikhan A, Ibrahimi OA, Eisen DB: Congenital melanocytic nevi:
Nicht-Mosaik-Erkrankungen ermöglicht außerdem, das where are we now? Part I. Clinical presentation, epidemiology,
Wiederholungsrisiko innerhalb einer Familie abzu- pathogenesis, histology, malignant transformation, and neurocutaneous
melanosis. J Am Acad Dermatol 2012; 67: 495. e1–17; quiz 512–4.
schätzen.
25. Kinsler VA, Boccara O, Fraitag S, Torrelo A, Vabres P, Diociauti A:
Mosaic abnormalities of the skin—review and guidelines from the
European Reference Network for rare skin diseases (ERN-Skin).
Interessenkonflikt ´ Br J Dermatol 2019; doi: 10.1111/bjd.17924 [Epub ahead of print].
Die Autoren erklären, dass kein Interessenkonflikt besteht.
26. Wang SM, Hsieh YJ, Chang KM, Tsai HL, Chen CP:
Manuskriptdaten Schimmelpenning syndrome: a case report and literature review.
eingereicht: 23. 4. 2019, revidierte Fassung angenommen: 28. 11. 2019 Pediatr Neonatol 2014; 55: 487–90.
27. Ardinger HH, Horii KA, Begleiter ML: Expanding the phenotype
Literatur of oculoectodermal syndrome: possible relationship to
1. larke CM, Edwards JH, Smallpeice V: 21-trisomy/normal mosaicism in encephalocraniocutaneous lipomatosis. Am J Med Genet A 2007;
an intelligent child with some mongoloid characters. Lancet 1961; 1: 143A: 2959–62.
1028–30. 28. Boppudi S, Bögershausen N, Hove HB, et al.: Specific mosaic KRAS
2. Rohlin A, Wernersson J, Engwall Y, Wiklund L, Björk J, Nordling M: mutations affecting codon 146 cause oculoectodermal syndrome and
Parallel sequencing used in detection of mosaic mutations: compari- encephalocraniocutaneous lipomatosis. Clin Genet 2016; 90: 334–42.
son with four diagnostic DNA screening techniques. Hum Mutat 2009; 29. Moog U: Encephalocraniocutaneous lipomatosis. J Med Genet 2009;
30: 1012–20. 46: 721–9.
124 Deutsches Ärzteblatt | Jg. 117 | Heft 8 | 21. Februar 2020MEDIZIN
30. Bennett JT, Tan TY, Alcantara D, et al.: Mosaic activating mutations 38. Leoni C, Gullo G, Resta N, et al.: First evidence of a therapeutic effect
in FGFR1 cause encephalocraniocutaneous lipomatosis. Am J Hum of miransertib in a teenager with Proteus syndrome and ovarian carci-
Genet 2016; 98: 579–87. noma. Am J Med Genet A 2019; 179: 1319–24.
31. Valera ET, McConechy MK, Gayden T, et al.: Methylome analysis and 39. Rouillé T, Aractingi S, Kadlub N, et al.: Local inhibition of MEK/Akt
whole-exome sequencing reveal that brain tumors associated with prevents cellular growth in human congenital melanocytic nevi. J Invest
encephalocraniocutaneous lipomatosis are midline pilocytic astrocyto- Dermatol 2019; 139: 2004–15. e13.
mas. Acta Neuropathol 2018; 136: 657–60. 40. Stosser MB, Lindy AS, Butler E, et al.: High frequency of mosaic
32. Shirley MD, Tang H, Gallione CJ, et al.: Sturge-Weber syndrome and pathogenic variants in genes causing epilepsy-related neurodevelop-
port-wine stains caused by somatic mutation in GNAQ. N Engl J Med mental disorders. Genet Med 2018; 20: 403–10.
2013; 368: 1971–9.
33. Al-Olabi L, Polubothu S, Dowsett K, et al.: Mosaic RAS/MAPK Anschrift für die Verfasser
variants cause sporadic vascular malformations which respond to Prof. Dr. Dr. med. Ute Moog
targeted therapy. J Clin Invest 2018; 128: 1496–508. Institut für Humangenetik
Universitätsklinikum Heidelberg
34. Eerola I, Boon LM, Mulliken JB, et al.: Capillary malformation-arterio- Im Neuenheimer Feld 440, 69120 Heidelberg
venous malformation, a new clinical and genetic disorder caused by ute.moog@med.uni-heidelberg.de
RASA1 mutations. Am J Hum Genet 2003; 73: 1240–9.
35. Qin L, Wang J, Tian X, et al.: Detection and quantification of mosaic Zitierweise
mutations in disease genes by next-generation sequencing. J Mol Moog U, Felbor U, Has C, Zirn B: Disorders caused by genetic mosaicism.
Diagn 2016; 18: 446–53. Dtsch Arztebl Int 2020; 117: 119–25. DOI: 10.3238/arztebl.2020.0119
36. Keppler-Noreuil KM, Parker VE, Darling TN, Martinez-Agosto JA:
Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & ►Die englische Version des Artikels ist online abrufbar unter:
therapeutic strategies. Am J Med Genet C Semin Med Genet 2016; www.aerzteblatt-international.de
172: 402–21. Zusatzmaterial
37. Venot Q, Blanc T, Rabia SH, et al.: Targeted therapy in patients with Mit „e“ gekennzeichnete Literatur:
PIK3CA-related overgrowth syndrome. Nature 2018; 558: 540–6. www.aerzteblatt.de/lit0820 oder über QR-Code
KLINISCHER SCHNAPPSCHUSS
Thrombophlebitis migrans
Ein 90-jähriger Mann stellte sich mit akut aufgetretenen
multiplen, schmerzhaften, tastbaren, flächigen, zum Teil
strangförmigen Thrombophlebitiden an Ober- und Unter-
armen vor. Blutentnahmen oder eine Anlage von peripher-
venösen Verweilkanülen waren innerhalb der letzten
14 Tage nicht erfolgt. Wenige Wochen zuvor war bei dem
Patienten eine Haarzellleukämie (ein niedrigmalignes
B-Zell-Non-Hodgkin-Lymphom) diagnostiziert und eine
zytostatische Therapie mit Cladribin begonnen worden.
Aufgrund der Anamnese und des charakteristischen
klinischen Bildes wurde die Diagnose Thrombophlebitis
migrans gestellt. Die Thrombophlebitis migrans, auch
Trousseau-Syndrom genannt, ist ein fakultativ paraneo-
plastisches Syndrom und kann somit ein Anzeichen für
Tumorerkrankungen darstellen. Ätiopathogenetisch liegt
der Thrombophlebitis migrans wahrscheinlich eine intra-
vasale Koagulation durch prokoagulatorische Faktoren der
Tumorerkrankung zugrunde. Bei ungewöhnlichen Throm-
bophlebitiden ohne fassbare Ursache sollte daher immer auch an ein okkultes Tumorleiden gedacht werden. Thera-
peutisch können neben einer Kompressionstherapie und der Gabe von Antiphlogistika auch Blutgerinnungshemmer
wie niedermolekulare Heparine verabreicht werden. In unserem Fall führte eine Behandlung mit Fondaparinux, einem
selektiven Faktor-Xa-Hemmer, zu einer schnellen Abheilung aller Läsionen.
Dr. med. Bijan Koushk-Jalali, Dr. med. Christian Tigges, Prof. Dr. med. Alexander Kreuter,
Klinik für Dermatologie, Venerologie und Allergologie, HELIOS St. Elisabeth Klinik Oberhausen, Universität Witten-Herdecke,
alexander.kreuter@helios-gesundheit.de
Interessenkonflikt: Die Autoren erklären, dass kein Interessenkonflikt besteht.
Zitierweise: Koushk-Jalali B, Tigges C, Kreuter A: Thrombophlebitis migrans. Dtsch Arztebl Int 2020; 117: 125. DOI: 10.3238/arztebl.2020.0125
►Vergrößerte Abbildung und englische Übersetzung unter: www.aerzteblatt.de
Deutsches Ärzteblatt | Jg. 117 | Heft 8 | 21. Februar 2020 125MEDIZIN
Zusatzmaterial zu:
Erkrankungen durch genetische Mosaike
Ute Moog, Ute Felbor, Cristina Has, Birgit Zirn
Dtsch Arztebl Int 2020; 117: 119–25. DOI: 10.3238/arztebl.2020.0119
eLiteratur
e1. Happle R: Kutane Mosaike: Muster und molekulare Mechanismen.
Dtsch Arztebl 2004; 101: A-1886.
e2. Happle R: Mosaicism in human skin. Understanding the patterns
and mechanisms. Arch Dermatol 1993; 129: 1460–70.
e3. Mirzaa G, Conway R, Graham JM Jr, et al.: PIK3CA-Related Seg-
mental Overgrowth. In: Adam MP, Ardinger HH, Pagon RA, et al.,
(eds.): GeneReviews. Seattle (WA): University of Washington 2013;
www.ncbi.nlm.nih.gov/books/NBK153722/ (last accessed on 28
January 2020).
e4. Jamuar SS, Lam AT, Kircher M, et al.: Somatic mutations in cerebral
cortical malformations. N Engl J Med 2014; 371: 733–43.
I Deutsches Ärzteblatt | Jg. 117 | Heft 8 | 21. Februar 2020 | ZusatzmaterialSie können auch lesen

















































