Induktion des immunogenen Zelltods bei adhärenten Tumorzellen durch gezielte Anreicherung von Mitoxantron durch magnetische Eisenoxidnanopartikel ...
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Induktion des immunogenen Zelltods bei adhärenten
Tumorzellen durch gezielte Anreicherung von
Mitoxantron durch magnetische
Eisenoxidnanopartikel als Transportsystem
Hals-Nasen-Ohren-Klinik, Kopf- und Halschirurgie
Sektion für Experimentelle Onkologie und Nanomedizin (SEON)
Universitätsklinikum Erlangen
Medizinischen Fakultät
der Friedrich-Alexander-Universität
Erlangen-Nürnberg
zur
Erlangung des Doktorgrades Dr. med.
vorgelegt von
Teresa Ratschker
1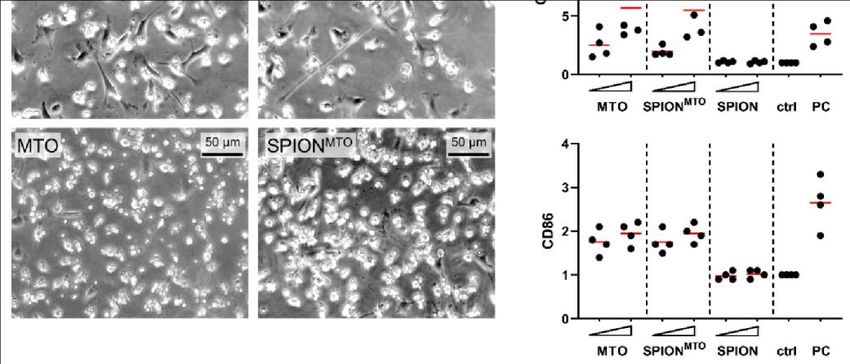
Als Dissertation genehmigt
von der Medizinischen Fakultät
der Friedrich-Alexander-Universität Erlangen-Nürnberg
Vorsitzender des Promotionsorgans: Prof. Dr. med. Markus F. Neurath
Gutachter: Prof. Dr. Christoph Alexiou
Gutachter: Prof. Dr. Udo Gaipl
Tag der mündlichen Prüfung: 29.06.2021
Für Felix
2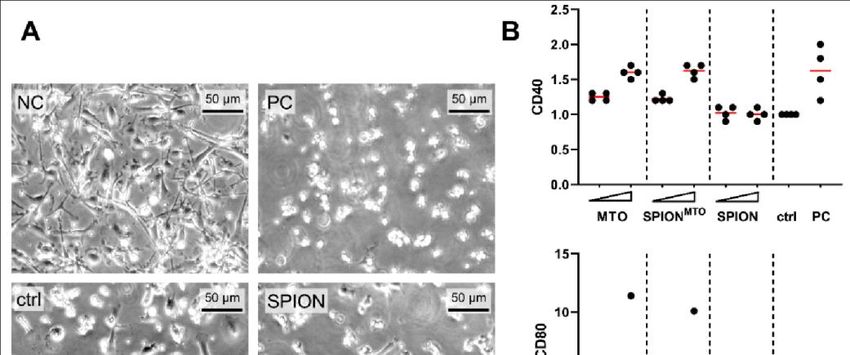
Inhaltsverzeichnis
1. Zusammenfassung ........................................................................................................ 1
2. Einordnung in den wissenschaftlichen Kontext ............................................................ 3
2.1 Notwendigkeit der Chemotherapie in der Behandlung solider Tumore ............................ 3
2.2 Tumorentstehung ............................................................................................................... 3
2.3 Wirkmechanismus von Zytostatika ..................................................................................... 4
2.4 Formen des Zelltodes .......................................................................................................... 5
2.4.1 Nekrose ................................................................................................................. 5
2.4.2 Apoptose ............................................................................................................... 5
2.4.3 Immunogener Zelltod ........................................................................................... 6
2.5 Rolle des Immunsystems .................................................................................................... 6
2.5.1 Immunologische Kontrolle der Tumorentstehung................................................ 7
2.5.2 Checkpoint-Inhibitoren ......................................................................................... 8
2.5.3 Zytokinbasierte Krebsimmuntherapie .................................................................. 9
2.5.4 T-Zell basierte Krebsimmuntherapie .................................................................. 10
2.5.5 Impfung gegen Tumore....................................................................................... 10
2.5.6 ICD-induzierende Chemotherapeutika ............................................................... 13
2.6 Nebenwirkungen der systemischen Chemotherapie........................................................ 13
2.7 Gezielte Krebstherapie...................................................................................................... 14
2.7.1 Passiver Medikamententransport ...................................................................... 14
2.7.2 Aktiver Medikamententransport ........................................................................ 15
2.8 Magnetisches Drug Targeting .......................................................................................... 16
2.8.1 Synthese der SPIONs und Beladung mit Chemotherapeutikum ......................... 17
2.8.2 Magnetische Akkumulation des Wirkstoffs im Tumor........................................ 17
2.9 Zielgerichtete Chemotherapie mit Induktion einer Immunantwort................................. 18
3. Publikationsliste .......................................................................................................... 20
Mitoxantrone-Loaded Nanoparticles for Magnetically Controlled Tumor Therapy–Induction of
Tumor Cell Death, Release of Danger Signals and Activation of Immune Cells.......................... 21
1. Introduction ........................................................................................................................ 22
2. Materials and Methods ....................................................................................................... 23
2.1. Cell Lines and Culture Conditions .................................................................................... 24
2.2. Synthesis and Characterization of Superparamagnetic IronOxide Nanoparticles(SPIONs)24
2.3. Endotoxin Content ........................................................................................................... 24
2.4. Plasma Coagulation .......................................................................................................... 25
2.5. Platelet Aggregation ........................................................................................................ 25
3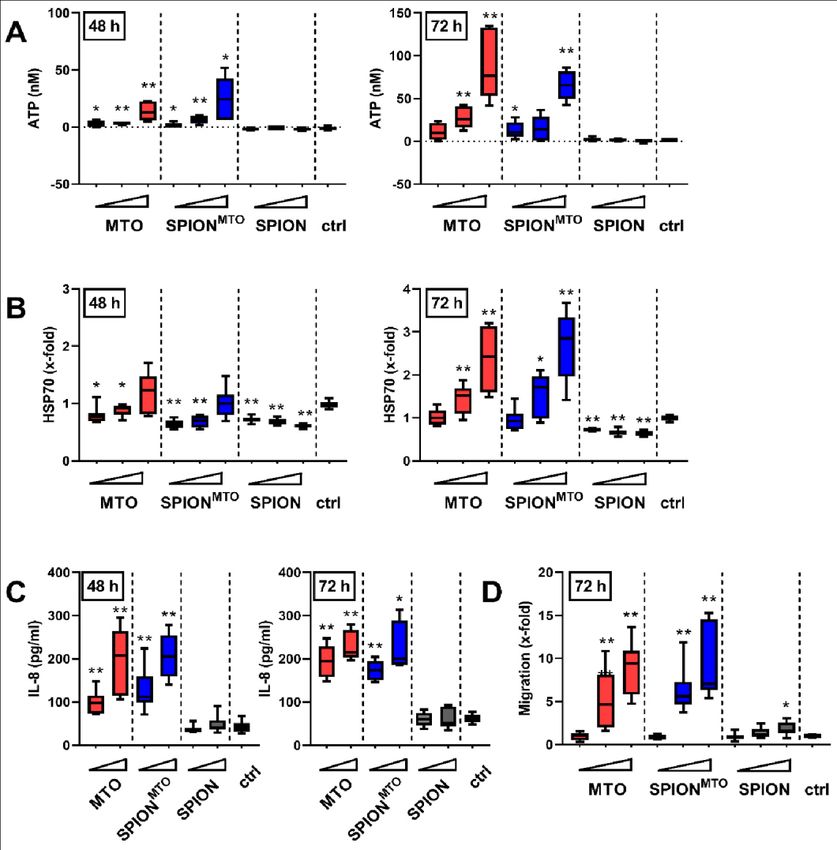
2.6. Hemolysis ......................................................................................................................... 25
2.7. Magnetic Accumulation of SPIONMTO ............................................................................... 26
2.8. Determination of Cell Proliferation.................................................................................. 26
2.9. Determination of Cell Cycle and Cell Death ..................................................................... 26
2.10. Flow Cytometry .............................................................................................................. 27
2.11. ATP Release .................................................................................................................... 27
2.12. Heat Shock Protein 70 (HSP70) Release ........................................................................ 27
2.13. Chemotaxis..................................................................................................................... 28
2.14. Generation of Human Immature Dendritic Cells (iDCs) ................................................. 28
2.15. Maturation of Human Dendritic Cells with Supernatants of HT-29 Tumor Cells .......... 28
2.16. Cytokine Analysis ........................................................................................................... 29
2.17. Data Analysis and Statistics............................................................................................ 29
3. Results ................................................................................................................................. 29
3.1. Biocompatibility ............................................................................................................... 29
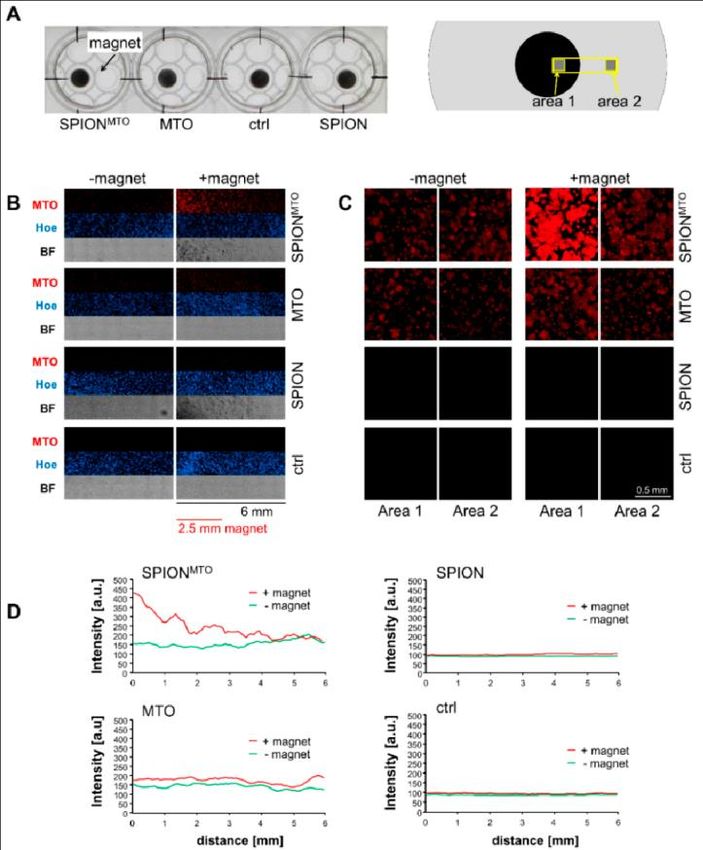
3.2. Magnetic Accumulation of SPIONMTO ............................................................................... 31
3.3. Drug Uptake and Cell Death Induction............................................................................. 32
3.4. Release of Damage-Associated Molecular Patterns (DAMPs) and Activation of Immune
Cells…………………….…………………………………… ……………………………………………………………………….34
4. Discussion............................................................................................................................ 36
References ........................................................................................................................ 39
4. Abkürzungsverzeichnis................................................................................................ 43
5. Literaturverzeichnis .................................................................................................... 44
4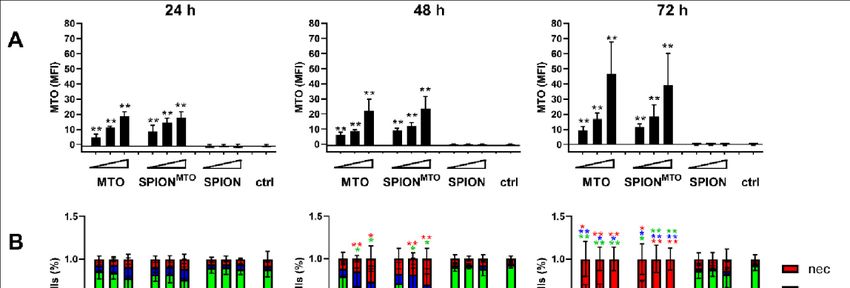
1. Zusammenfassung
Hintergrund und Ziele: Mitoxantron (MTO) gilt als typischer Induktor des immuno-
genen Zelltodes (ICD), der die Freisetzung von inflammatorischen Gefahrensignalen
(„Damage-associated molecular patterns“ – DAMPs) bei sterbenden Tumorzellen
auslöst. Wenn es als Chemotherapeutikum intravenös appliziert wird, kann es jedoch
aufgrund seiner systemischen Verteilung schwere Nebenwirkungen im gesunden Gewebe
hervorrufen. Die Bindung an Eisenoxidnanopartikel (SPIONs) als Transportersystem
(SPIONMTO) ermöglicht die magnetische Anreicherung der Substanz. Ziel dieser
Doktorarbeit war es, die Wirksamkeit von freiem MTO mit der von SPION -gebundenem
MTO auf adhärente Kolonkarzinomzellen zu vergleichen und die freigesetzten
Botenstoffe auf ihre Fähigkeit zur Induktion einer Immunantwort hin zu untersuchen. Zur
Vorbereitung einer in vivo Anwendung wurde zudem die Hämokompatibilität der Partikel
untersucht und die magnetische Akkumulierbarkeit der Nanopartikel überprüft.
Material und Methoden: Adhärente Kolonkarzinomzellen (HT-29) wurden im 2D-
Modell für eine definierte Zeit (24h, 48h oder 72h) mit SPIONs, MTO oder SPIONMTO
behandelt. Dann wurden die Zellen hinsichtlich ihrer exprimierten Oberflächenproteine,
intrazellulären ATP-Speicher, Plasmamembranpermeabilität und des Zellzyklusstatus
mittels Multiparameterfärbungen in der Durchflusszytometrie untersucht. Im Überstand
der sterbenden Zellen wurde die Konzentration von ATP, HSP70 und Interleukin-8
mittels Chemilumineszenz, ELISA und Cytometric Bead Array gemessen. Die chemo-
taktische Aktivität des Überstandes auf THP-1 Monozyten wurde im Transwell System
untersucht. Die Überstände wurden anschließend auf dendritische Zellen (DC) gegeben.
Mithilfe von Antikörperfärbungen der Aktivierungsmarker wurde untersucht, ob eine
Reifung der DCs induziert wird. Außerdem wurden mittels eines extern angelegten
Magnetfeldes die Partikel in einem Bereich der Wells angereichert und die intrazelluläre
Konzentration von MTO in Bereichen mit starkem und schwachem Magnetfeld mittels
Fluoreszenzmikroskopie untersucht. Parallel dazu wurde die Hämokompatibilität (Ery-
throzytenverträglichkeit, Plasmakoagulation, Thrombozytenaktivierung) der Partikel
überprüft.
Ergebnisse und Beobachtungen: Es konnte gezeigt werden, dass an SPIONs
gebundenes MTO (SPIONMTO) genauso wirkt wie freies MTO. Die reinen SPIONs
1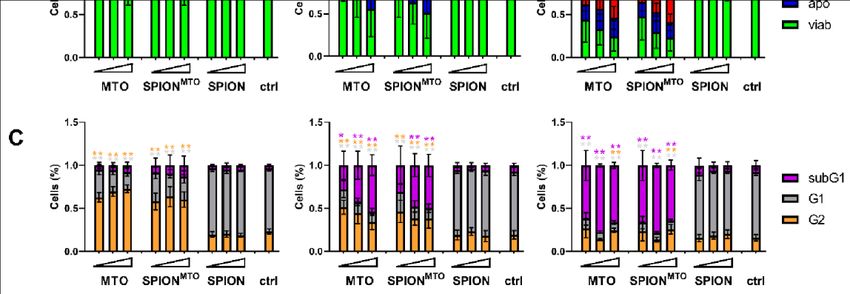
hingegen haben keinen Einfluss auf das Zellwachstum oder den Zellzyklus. Bei freiem
MTO und SPIONMTO trat nach 24 h ein Zellzyklusarrest ein und im weiteren Verlauf
gingen die Tumorzellen in die Apoptose. Proportional zum Anteil der sterbenden/toten
Zellen stieg die Konzentration der sekretierten DAMPs (ATP, HSP70, Interleuktin-8) an.
Die Reifung der dendritischen Zellen wurde angeregt und Monozyten angelockt. Die
Partikel ließen sich durch ein externes Magnetfeld anziehen, wodurch das
Chemotherapeutikum stark angereichert wurde und lokal seine Wirkung entfaltete.
Außerdem konnte gezeigt werden, dass in den untersuchten Konzentrationen mit keiner
Hämolyse, Thrombenbildung oder Plättchenaktivierung im Blut zu rechnen ist.
Schlussfolgerungen: Es ist möglich, MTO an SPIONs zu binden und die toxische
Substanz so gezielt magnetisch anzureichern. Die Wirkung des MTO wird dabei nicht
eingeschränkt und es werden DAMPs freigesetzt, die eine Reifung von dendritischen
Zellen auslösen und Monozyten anlocken. SPIONMTO kann in den untersuchten
Konzentrationen intravasal appliziert werden, da es weder eine Hämolyse auslöst, noch
die primäre oder sekundäre Blutgerinnung negativ beeinflusst. In weiteren Experimenten
muss überprüft werden, ob die Immunzell-Aktivierung ausreicht, um Tumorzellen für das
Immunsystem als Gefahr sichtbar zu machen und so eine ausreichende Antitumor-
Reaktion zu erzeugen.
22. Einordnung in den wissenschaftlichen Kontext
2.1 Notwendigkeit der Chemotherapie in der Behandlung solider Tumore
Krebserkrankungen sind in Deutschland mit 230.031 Todesfällen allein 2018 die dritt-
häufigste Todesursache (Statistisches Bundesamt (Destatis), Stand: 16.09.2020). Ein
Großteil der Krebserkrankungen sind solide Tumore, die in vielen Fällen operativ entfernt
werden können.
Daneben gibt es die Möglichkeit, mit ionisierender Strahlung die Tumorzellen an einem
definierten Ort zu schädigen. Dies wird vor allem bei schlecht operablen Gebieten oder
ergänzend zur Operation durchgeführt, um die Tumorlast zu verkleinern und das
Rezidivrisiko zu senken (Sebag-Montefiore et al. 2009). Meist wird von außen mit
ionisierender Strahlung eine maximal tolerable Strahlendosis in das Tumorgewebe
gegeben. In einigen Fällen werden auch radioaktiv zerfallende α-Strahler als Seeds direkt
im Gewebe platziert und schädigen so von innen vor Ort das Tumorbett (Fitch et al. 2006).
Die dritte große Säule in der Behandlung von Karzinomen ist die Chemotherapie,
besonders wenn Tumorzellen bereits im Körper gestreut haben, der Tumor nicht resek-
tabel ist oder postoperativ Tumorzellen im Körper verbleiben (R1 Stadium), sowie in
Rezidivsituationen (S3-Leitlinie Kolorektales Karzinom). Durch eine Zytostatika-
Therapie kann bei vorliegenden Lymphknoten- (N1) oder anderen Metastasen (M1) das
Gesamtüberleben verbessert werden (Gill et al. 2004).
2.2 Tumorentstehung
Die Gemeinsamkeit aller Neoplasien ist eine mutierte DNA. Maligne Neoplasien
zeichnen sich vor allem durch Mutationen in Protoonkogenen und Tumorsupressorgenen
aus. Durch die Inaktivierung von Tumorsupressorgenen und die Aktivierung von Proto-
onkogenen teilt sich die Zelle schneller und unkontrolliert, da Regulationskomponenten
der DNA-Synthese übergangen werden (Moch et al. 2012a).
In diesem veränderten Zellzyklus ist es der Zelle nicht mehr möglich, neue Mutationen
zu verhindern, da die notwendigen Reparaturmechanismen ebenfalls ausgefallen sind.
Die entartete Zelle akkumuliert weitere Mutationen (Anandakrishnan et al. 2019). Je
entarteter die Zelle, desto aggressiver und schneller wächst der Tumor.
32.3 Wirkmechanismus von Zytostatika
Chemotherapeutika wirken auf schnell teilende Zellen, daher wirken sie besonders
effizient bei aggressiven Tumoren wie Lymphomen (Kriegsmann et al. 2018). Die
schnelle und unkontrollierte Replikation wird zum Angriffspunkt der Zytostatika. Diese
verhindern die Zellteilung, indem sie die DNA direkt schädigen oder wichtige Schritte
der Replikation hemmen und so die Zelle zerstören (Karow und Lang-Roth 2018a). Man
unterscheidet Zytostatika nach ihrem Wirkmechanismus und ihrer Molekül-Gruppe.
Antimetabolite wie 6-Mercaptopurin, 5-Fluorouracil (5-FU) oder Metotrexat werden als
Purin- oder Pyrimidinanaloga während der Replikation im Tochterstrang verbaut und
führen so zum Strangabbruch (Lansiaux 2011). Durch die Hemmung der DNA-Polyme-
rase verhindert Cytarabin die Replikation des Genoms in der Interphase (Rechkoblit et al.
2019).
Eine weitere wichtige Gruppe von Zytostatika sind die Alkylantien. Sie fügen Alkyl-
gruppen in die Doppelhelix-Struktur der DNA ein und verhindern so die Replikation
(Scheulen et al. 2003b). Platinderivate wie Cisplatin wirken ähnlich. Sie erzeugen
kovalente Interstrang-Quervernetzungen.
Mitoxantron (MTO) aus der Gruppe der Antrazykline wirkt, indem es einen ternären
Komplex zwischen der DNA-Topoisomerase und der doppelsträngigen DNA stabilisiert
und zusätzlich eine Interkalation in die DNA stattfindet. Jüngste Hinweise legen nahe,
dass MTO auch durch Hemmung der PIM1-Kinase-Aktivität, durch Induktion der
Bildung freier Sauerstoffradikale und durch Beeinflussung des Eisenstoffwechsels zyto-
toxisch wirkt (Shaul et al. 2015).
Topoisomerase-Inhibitoren müssen allerdings nicht direkt mit der DNA interagieren.
Auch die alleinige Hemmung der Topoisomerase ist bereits zytotoxisch, da die konden-
sierte DNA nicht mehr entspiralisiert werden kann. So können andere Enzyme wie z.B.
die DNA-Polymerase oder die RNA-Polymerase nicht an der DNA binden, wodurch die
Replikation und auch die Proteinbiosynthese verhindert wird (Scheulen et al. 2003a).
Einen ganz anderen Ansatz verfolgen Mitosehemmstoffe wie Taxane oder Vinka-
alkaloide, die nicht die Replikation der DNA-Stränge verhindern, sondern die korrekte
Trennung der Chromatiden unterbinden, indem der dafür benötigte Spindelapparat aus
Mikrotubuli nicht polymerisiert (bei Vinkaalkaloiden) bzw. depolymerisiert (bei
4Taxanen) werden kann. Die Zelle verbleibt in der Metaphase, kann die Mitose nicht been-
den und sich somit nicht teilen (Zhao et al. 2005).
2.4 Formen des Zelltodes
2.4.1 Nekrose
Werden Zellen starken physikalischen Veränderungen wie Hitze, Kälte oder mechani-
schen Kräften ausgesetzt, verlieren sie die Integrität ihrer Plasmamembran und platzen.
Dabei werden in der sogenannten primären Nekrose die intrazellulären Bestandteile unge-
ordnet freigesetzt und lösen im Gewebe eine Entzündungsreaktion aus, die auch
umliegende gesunde Zellen schädigen kann (Biermann et al. 2016, Golstein und Kroemer
2007).
Auch unter dem Einfluss der verschiedenen Chemotherapeutika kann es zum Verlust der
Plasmamembranintegrität kommen. Durch die Zytostatika wird das Genom bei der
Replikation zerstört und die Zelle kann keine ausreichende Proteinbiosynthese mehr
betreiben, um ihre Homöostase aufrecht zu erhalten. Die Zelle schwillt an und die
Plasmamembrane wird durchlässig. Bei dieser sogenannten sekundären Nekrose werden
ebenfalls intrazelluläre Bestandteile ungeordnet freigesetzt und lösen im Gewebe eine
überschießende Immunantwort aus (Tesniere et al. 2008, Silva 2010).
2.4.2 Apoptose
Davon zu unterscheiden ist der programmierte Zelltod, die sogenannte Apoptose (Kerr et
al. 1972). Für den Organismus ist es von entscheidender Bedeutung, dass nicht mehr
notwendige oder funktionsunfähige Zellen abgebaut werden können (Munoz et al. 2017).
In der Embryogenese beispielsweise werden gezielt Blutgefäße im Augapfel, die für die
Entwicklung und Versorgung der Linse nötig waren, wieder abgebaut, damit der
Glaskörper lichtdurchlässig wird (Bernoulli 1949).
Der Organismus entfernt außerdem alte oder beschädigte Zellen. Werden Zellschäden
rechtzeitig erkannt oder sind nicht so unmittelbar, dass sie zur Nekrose führen, geht die
Zelle geordnet zu Grunde (Elmore 2007). Für die Apoptose gibt es mehrere Auslöser wie
eine DNA-Schädigung, zellulärer Stress, fehlende Wachstumsfaktoren, Hypoxie oder die
Induktion mittels Botenstoffen oder Rezeptoren von außen. Die verschiedenen
Mechanismen münden alle in der Aktivierung von Caspasen, der Freisetzung von
Cytochrom C aus den Mitochondrien, der Exposition von Phosphatidylserin auf der
5äußeren Plasmamembrane und der Kondensation und Fragmentierung intranukleärer
DNA (De et al. 2018). Die Zelle schrumpft, wobei die Granularität durch kondensiertes
Chromatin steigt, und sich aus der Plasmamembrane kleine Blasen mit komprimierter
DNA und Zytoplasma, sogenannte Apoptosekörperchen, abstülpen (Elmore 2007). Die
Zelle zerfällt in Fragmente, die auf ihrer Oberfläche Fresssignale für Phagozyten tragen
und diese so anlocken. Das verbleibende Zellgeröll wird von Makrophagen abgeräumt,
was eine überschießende Immunreaktion verhindert (Munoz et al. 2017).
2.4.3 Immunogener Zelltod
Neben diesen beiden großen Gruppen gibt es noch weitere Unterformen des Zelltodes.
Einer davon ist der sogenannte immunogene Zelltod (Immunogenic cell death, kurz ICD).
Er kann vor allem bei Tumorzellen unter Strahlenbehandlung, photodynamischer
Therapie oder Chemotherapie mit Anthrazyklinen beobachtet werden (Tesniere et al.
2008, Kepp et al. 2014). ICD beschreibt einen programmierten Zelltod, der durch frei-
gesetzte Gefahrensignale, sogenannte „Damage associated molecular Patterns“
(DAMPs), eine Interaktion mit dem adaptiven Immunsystem initiiert (Galluzzi et al.
2020). Infolgedessen werden gezielt Immunzellen angelockt, Tumorantigene erkannt und
Tumorzellen attackiert (Hodge et al. 2013, Yatim et al. 2017). Charakteristischerweise
erfolgt die zeitlich koordinierte Exprimierung von Calreticulin (CRT) (Garg et al. 2012)
auf der Plasmamembranoberfläche sowie die Freisetzung von DAMPs wie das High
mobility group box protein B1 (HMGB1), Adenosintriphosphat (ATP) oder das Heat
shock protein 70 kDa (HSP70) (Krysko et al. 2012). Diese freigesetzten DAMPs binden
ihren zugehörigen Mustererkennungs-Rezeptor (Pattern recognition receptor) auf
Immunzellen und locken so Antigen-präsentierende Zellen (APCs) an. ATP beispiels-
weise bindet an purinerge P2X7 Rezeptoren auf dendritischen Zellen, wo es die NALP3-
ASC-Inflammasomen aktiviert und so die Freisetzung von IL-1β auslöst, sowie an P2Y2
Rezeptoren, was die Monozyten-Migration stimuliert (Krysko et al. 2012).
2.5 Rolle des Immunsystems
Wie aus den verschiedenen Zelltodformen schon ersichtlich, spielt das Immunsystem eine
entscheidende Rolle in der Erkennung und Bekämpfung von Tumoren (Showalter et al.
2017). Bei Patienten mit eingeschränktem Immunsystem, z.B. durch eine HIV-Infektion,
treten Tumore wie das Karposisarkom, das normalerweise vom Immunsystem verhindert
wird, vermehrt auf (Robey und Bower 2015).
6Umgekehrt zeigt sich bei klinisch relevanten soliden Tumoren häufig eine verminderte
Leukozyteninfiltration im Gewebe (Kather et al. 2018). So spricht man bei Tumoren mit
einer geringen Anzahl von infiltrierenden T-Lymphozyten von immunologisch „kalten“
Tumoren mit einer schlechteren Prognose (van der Woude et al. 2017).
Das Zusammenspiel der unterschiedlichen Leukozyten wurde erst in den letzten
Jahrzehnten zunehmend entschlüsselt. Kommt es zu einer Freisetzung von Antigenen in
den Extrazellularraum in einer inflammatorischen Umgebung, wie zum Beispiel bei
einem Zelluntergang, werden diese Antigene von den ubiquitär vorkommenden
dendritischen Zellen (DC) aufgenommen, prozessiert und in den nächsten Lymphknoten
gebracht, wo sie naive T-Zellen primen. Mittels MHC-I-Komplex wird das spezifische
Antigen den zytotoxischen CD8+ T-Zellen, sowie mit dem MHC-II-Komplex den CD4+
T-Helfer-Zellen präsentiert. Dendritische Zellen aktivieren somit als APCs Lymphozyten
(Lüllmann-Rauch 2009). Die aktivierten T-Zellen wandern in den Blutkreislauf und über
das Kapillarsystem wieder zurück an den Ursprung des Antigens, dem Bereich des
Zelluntergangs. Entzündungsreaktionen unterstützen durch freigesetzte Zytokine die
Migration der T-Zellen ins Gewebe. Wird eine Zelle mit entsprechendem Antigen durch
den T-Zellrezeptor erkannt, induziert die T-Zelle die Apoptose der infizierten Zelle.
2.5.1 Immunologische Kontrolle der Tumorentstehung
Neben der Erkennung und Eliminierung von körperfremden Pathogenen besitzt das
Immunsystem auch die Möglichkeit, veränderte körpereigene Zellen zu erkennen
(Waldman et al. 2020). Jede Zelle präsentiert auf ihrer Oberfläche mittels des MHC-I-
Komplexes zytoplasmatische Proteinfragmente. Ist die DNA durch spontane Mutationen
oder durch eine Virusinfektion der Zelle beschädigt, werden veränderte Proteine
synthetisiert und im MHC-I-Komplex präsentiert.
Kommt es zur Freisetzung dieser Mutations-assoziierten Proteine, nehmen die
dendritischen Zellen diese als Antigene auf und präsentieren sie im zugehörigen
Lymphknoten naiven T-Zellen. Die Erkennung erfolgt mittels des T-Zell-Rezeptors
(TCR) der T-Zelle und dem MHC-Rezeptor der APC, auf dem das Antigen präsentiert
wird. Neben dieser stimulierenden Interaktion laufen gleichzeitig hemmende Kontakte,
wie eine Interaktion der Rezeptoren CD80/CD86 der APC und CTLA-4 der T-Zelle ab.
Überwiegen die stimulierenden Faktoren, so kommt es zur Aktivierung der zytotoxischen
T-Zellen, welche zurück zum Tumor wandern (Chen und Mellman 2013, Makkouk und
Weiner 2015). Die aktivierten CD8+ T-Lymphozyten erkennen und binden mutierte
7Zellen anhand ihrer auf den MHC-I -Komplex gebundenen Tumorantigene und treiben
so die Tumorzellen mittels zytotoxischer Substanzen, wie Perforinen und Granzymen
(Kitamura et al. 2019), oder rezeptorvermittelt in die Apoptose (Seremet et al. 2011),
wodurch erneut Antigene freigesetzt werden. Funktioniert dieser sogenannte Krebs-
Immunzell-Zyklus, wird der Tumor eliminiert (Chen und Mellman 2013). Die
Überwachung von mutierten Zellen durch das Immunsystem wird Tumorsurveillance
genannt (Dunn et al. 2002).
Unter diesem Selektionsdruck (sog. immunoediting) proliferieren vermehrt Tumorzellen,
die dem Angriff des Immunsystems entgehen können (Beatty und Gladney 2015). Dafür
produzieren sie beispielsweise weniger Antigene, exprimieren weniger MHC-I-
Komplexe (Khong et al. 2004), bilden Resistenzen gegen zytotoxische Botenstoffe,
gestalten ihre Mikroumgebung immunsuppressiv oder verhindern rezeptorvermittelt eine
ausreichende T-Zell-Antwort. Durch Überexpression von Programmed death Ligand 1
(PD-L1) beispielsweise interagieren Tumorzellen mit den PD-1 Rezeptoren auf
zytotoxischen CD8+ T-Lymphozyten und verhindern ihren Untergang (Gatalica et al.
2014). Außerdem werden T-Zellen dadurch inaktiviert und sogar zur Apoptose gedrängt
(Waldman et al. 2020).
2.5.2 Checkpoint-Inhibitoren
Mittels der Krebsimmuntherapie versucht man die Immunantwort gegen den Tumor
wieder zu verstärken. Checkpoint-Inhibitoren machen sich Antikörper zu Nutze, die PD-
1 auf Tumorzellen oder dessen Ligand PD-L1 auf T-Zellen binden. Dadurch wird die
hemmende Interaktion zwischen Tumor und T-Lymphozyt blockiert (He et al. 2016,
Cantara et al. 2019), so können die T-Lymphozyten wieder gegen die Tumorzellen
vorgehen (Wu et al. 2019). In Experimenten konnte gezeigt werden, dass PD-1
Inhibitoren als Co-Stimulanz die Krebsimmuntherapie verbessern können, indem sie die
T-Zell-Antwort verstärken (Zhu et al. 2017). Zurzeit sind mehrere Checkpoint-
Inhibitoren in der Klinik zugelassen, unter anderem die PD-1 Antikörper Nivolumab und
Pembrolizumab zur Therapie von Melanomen, nicht kleinzelligen Lungenkarzinomen
und Hodgkin Lymphomen, der PD-L1 Antikörper Avelumab zur Merkelzellkarzinom-
Therapie und der PD-L1 Antikörper Atezolizumab in der Therapie von
Urothelkarzinomen und nicht kleinzelligen Lungenkarzinomen (Heinzerling et al. 2019).
82.5.3 Zytokinbasierte Krebsimmuntherapie
Neben Checkpoint-Inhibitoren, die eine spezifische Schaltstelle der Immunantwort
beeinflussen, wird auch versucht, mit Lymphozyten-unterstützenden Zytokinen die
generelle Immunantwort zu steigern.
Hierfür werden Interleukine (IL), Interferone (INF) oder Leukozyten-stimulierende
Faktoren wie Granulozyten-stimulierender Faktor (G-CSF) und Granulozyten/Makro-
phagen-stimulierender Faktor (GM-CSF) eingesetzt (Riley et al. 2019).
Als Botenstoffe zwischen Leukozyten koordinieren Interleukine (IL) die Immunantwort.
IL-2 beispielsweise wird von T-Lymphozyten produziert und stimuliert T-, B- und
Natürliche Killer-Zellen (NK-Zellen) (Rassow 2006). Daher wird es im Rahmen von
klinischen Studien aktuell in der Therapie des malignen Melanoms eingesetzt (S3-Leit-
linie zur Diagnostik, Therapie und Nachsorge des Melanoms). In Studien konnte gezeigt
werden, dass eine intratumorale Injektion von IL-2 in metastasierte Melanome das
Gesamtüberleben verlängert (Weide et al. 2010).
Auch Interferone (INF) können als Botenstoffe des Immunsystems Abwehrreaktionen
verstärken und werden daher in mehreren Tumortherapien, wie zum Beispiel in der
Behandlung des fortgeschrittenen Nierenzellkarzinoms eingesetzt (S3-Leitlinie
Diagnostik, Therapie und Nachsorge des Nierenzellkarzinoms). IFN-γ und GM-CSF sind
für die DC-Reifung und Makrophagenaktivierung von zentraler Bedeutung. DCs setzen
ihrerseits Zytokine wie IL-1β, IL-6, IL-12 oder TNF frei, die die Antworten der NK-
Zellen und T-Zellen unterstützen (Showalter et al. 2017).
Interferon α (IFN-α) führt bei allen Zellen zu einer vermehrten MHC-I-Expression und
somit zu einer besseren Identifikation von mutierten Zellen, sowie zu einer Aktivierung
von NK-Zellen (Baumann 2010). Es ist unter anderem in der Therapie von malignen
Melanomen (S3-Leitlinie zur Diagnostik, Therapie und Nachsorge des Melanoms),
chronisch myeloischen Leukämien (Leitlinie Chronisch Myeloische Leukämie),
Haarzell-Leukämien und einigen Non-Hodgkin-Lymphomen (Karow und Lang-Roth
2018c) zugelassen.
G-CSF und GM-CSF verursachen eine verstärkte Proliferation der namensgebenden
Zellreihen, sodass die generelle Immunreaktion verbessert wird. Insbesondere bei durch
Chemotherapie bedingten Neutropenien wird G-CSF standardmäßig eingesetzt (S3-
Leitlinie Supportive Therapie bei onkologischen PatientInnen) und soll bei sehr
aggressiven Schemata, wie dem eskaliertem BEACOPP beim Hodgkin Lymphom auch
9prophylaktisch verabreicht werden (S3-Leitlinie Diagnostik, Therapie und Nachsorge des
Hodgkin Lymphoms bei erwachsenen Patienten).
2.5.4 T-Zell basierte Krebsimmuntherapie
Wie schon beim Krebsimmunzell-Zyklus beschrieben, spielen die T-Zellen eine
entscheidende Rolle in der Eliminierung von Tumorzellen.
Bereits vor 40 Jahren gab es Ansätze, Patienten mit malignem Melanom mittels adoptiver
T-Zelltransfer (ATC)-Therapie zu behandeln (Rosenberg et al. 1988). Hierfür wurden aus
einer Krebsbiopsie isolierte Lymphozyten mittels IL-2-Behandlung in vitro vermehrt und
dann dem Patienten mit einem erneuten IL-2 Bolus wieder intravenös verabreicht
(Waldman et al. 2020).
Allerdings kann dieser Ansatz nur bei ausreichender T-Zell-Infiltration eingesetzt
werden. Immunologisch „kalte“ Tumore eignen sich nicht. Außerdem entgehen Tumore,
die auf Grund von geringer MHC-Expression nicht immunogen sind, weiterhin den
körpereigenen, wenn auch in vitro vermehrten, T-Zellen.
Eine Lösung dafür bietet die CAR T-Zell-Therapie, welche auch diese Tumorzellen
attackiert. In dieser Individualtherapie werden dem Patienten zuerst mittels Leukozyten-
apherese T-Zellen entnommen und unter Zytokinbehandlung, z.B. IL-2, in vitro klonal
vermehrt. Mittels z.B. viraler Vektoren wird dann eine Transduktion induziert, sodass die
T-Zellen chimäre Antigenrezeptoren (CAR) auf der Zelloberfläche bilden, die sich gegen
typische Tumorantigene richten. Allerdings sind diese veränderten T-Zell-Rezeptoren
nicht auf eine Antigenpräsentation mittels MHC-I-Komplex angewiesen, und erkennen
auch Tumorzellen mit verminderten MHC-Komplexen (Yu und Hua 2019).
Aktuell sind zwei Medikamente für die klinische Behandlung zugelassen,
Tisagenlecleucel in der Therapie der akuten B-Zell Leukämie und des diffus
großzelligen B-Zell-Lymphoms (Halford et al. 2020) und Axicabtagen-Ciloleucel bei
rezidiviertem oder refraktärem diffus großzelligem B-Zell-Lymphom und als
Drittlinientherapie bei primär mediastinalem großzelligem B-Zell-Lymphom (American
Association for Cancer Research 2018).
2.5.5 Impfung gegen Tumore
Eine Möglichkeit, das Immunsystem besser zu justieren und auf Krankheiten aufmerksam
zu machen, ist die aktive Immunisierung, bei der Antigene von Krankheitserregen in
geringer Dosis gezielt dem Immunsystem präsentiert werden, das daraufhin reagiert,
10Gedächtniszellen generiert und bei erneutem Kontakt das Antigen mittels
Antikörperbildung zügig eliminiert (Karow und Lang-Roth 2018b).
Diese „Wunderwaffe“ der Medizin wird seit über einem Jahrhundert sehr erfolgreich
gegen Infektionskrankheiten verwendet (WHO Europa 2018). Erst in jüngerer Zeit wurde
man auch auf den Nutzen gegenüber Tumorerkrankungen aufmerksam. Dabei ist die
prophylaktische von der therapeutischen Impfung zu unterscheiden. Erstere schützt den
Patienten vor einer Infektion durch einen onkogenen Virus, der in infizierten Zellen eine
Mutation induziert, und im weiteren Verlauf zu Krebs führen kann (Moch et al. 2012b).
Ein Paradebeispiel ist das humane Papilloma Virus (HPV), das maßgeblich an der
Entstehung vom Zervixkarzinomen und weiteren anogenitalen Tumoren beteiligt ist
(Tornesello et al. 2011). Groß angelegte Impfprogramme gegen die am häufigsten mit
Cervixkarzinomen assoziierten Subtypen HPV 16 und 18 konnten zu einer deutlichen
Abnahme der Infektionen und auch der konsekutiven Tumorinzidenz führen (WHO et al.
2017, Grubert 2003).
Ein weiteres Beispiel ist die gezielte Immunisierung gegen das Hepatitis B-Virus. Eine
chronische Hepatitis B-Infektion geht mit einem 60-fach erhöhten Risiko für ein
Hepatozelluläres Karzinom (HCC) einher (Herold 2019). So ist in Ländern mit hoher
Durchseuchungsrate auch die Prävalenz des HCC erhöht. In Impfprogrammen wird daher
versucht, dieser Tumorerkrankung mittels Primärprävention der zugrundeliegenden
Infektion vorzubeugen (Spearman und Sonderup 2014).
Wenn bereits neoplastische Zellen vorliegen und das Immunsystem mittels Impfung auf
diese schädlichen Zellen aufmerksam gemacht wird, spricht man von einer
therapeutischen Impfung gegen Tumore. Dafür werden meist typische Tumorantigene
verwendet. Viele Tumortypen zeichnen sich patientenübergreifend durch wieder-
kehrende Proteinkomplexe aus. Dies sind meist proliferationsfördernde und somit pro-
onkogene Proteine oder Beiprodukte, die bei typischen Mutationen in diesem Gewebe
entstehen. Beispielsweise wurde die Überexpression des epidermalen Wachstumsfaktor-
Rezeptors (EGF-R) durch Tumorzellen in über der Hälfte der Lungenkrebsfälle
beobachtet und gilt als ein Mechanismus dieser Krebsentstehung. CIMAvax-EGF, ein
therapeutischer Impfstoff für nicht-kleinzelligen Lungenkrebs, besteht aus einem
humanen rekombinanten epidermalen Wachstumsfaktor (EGF), der in der Lage ist, Anti-
körper gegen den autologen EGF zu induzieren, was zu einer Abnahme des EGF im
Serum und einer geringeren Interaktion zwischen EGF und EGF-R führt (Tagliamento et
11al. 2018). Die Krebszellen werden nicht direkt attackiert. Vielmehr wird durch den Entzug
des Wachstumsstimulus die überschießende Proliferation gehemmt. Aktuell befindet sich
der Wirkstoff noch in der Entwicklung.
Einen anderen Mechanismus verwendet der therapeutische Krebsimpfstoff Sipuleucel-T,
der in den USA für die Therapie des Prostatakarzinoms zugelassen ist. Bei einer
Immuntherapie mit Sipuleucel-T werden dem Patienten APCs mittels Leukapherese
entnommen, mit einem Fusionsprotein aus prostataspezifischer saurer Phosphatase (PSA)
und GM-CSF inkubiert und dann reinfundiert, um so eine Immunantwort gegen den
Tumor zu induzieren (Botrel et al. 2012). Da PSA fast ausschließlich von entarteten
Prostatazellen produziert wird, ist die Wirkung an gesundem Gewebe sehr gering.
Sipuleucel-T ist seit Mitte 2015 nicht mehr in Europa erhältlich und wird daher in der
aktuellen Leitlinie Prostatakarzinom nicht mehr empfohlen (S3-Leitlinie zur
Früherkennung, Diagnose und Therapie der verschiedenen Stadien des
Prostatakarzinoms).
Ein weiterer Impfstoff, der in der Krebsimmuntherapie verwendet wird, ist Bacillus-
Calmette–Guérin (BCG), ein attenuierter Mykobakterienstamm, der ursprünglich als
Tuberkuloseimpfstoff entwickelt wurde. Seit über 30 Jahren ist bekannt, dass eine
intravesikale Installation von BCG bei Blasenkarzinomen zu einer Remission führt und
so eine Zystektomie ersetzen kann (De Jager et al. 1991, Han et al. 2020). Die aktuelle
S3-Leitlinie Harnblasenkarzinom empfiehlt bei Patienten mit einem intermediate-risk
Stadium sowohl bei Erstdiagnose als auch in einer Rezidivsituation eine
Installationstherapie mit BCG (S3-Leitlinie Früherkennung, Diagnose, Therapie und
Nachsorge des Harnblasenkarzinoms). Zwar kann man bei dieser Therapie von keiner
Tumorimpfung im eigentlichen Sinne sprechen, da dem Patienten keine
tumorspezifischen Antigene appliziert werden, die eine adaptive Immunantwort auslösen.
Der genaue Wirkmechanismus ist Gegenstand aktueller Forschungen. Allerdings gehen
bisherige Erkenntnisse davon aus, dass die Antitumorwirkung von BCG durch ein
Zusammenspiel zwischen den direkten Auswirkungen der BCG-Infektion auf
Tumorzellen und der Immunantwort des Wirts hervorgerufen wird. Das Ergebnis dieser
Immunaktivierung ist eine verbesserte Erkennung und anschließende Zerstörung von
Tumorzellen durch zellvermittelte, unspezifische und spezifische Mechanismen (Fuge et
al. 2015).
122.5.6 ICD-induzierende Chemotherapeutika
Auch ICD-induzierende Medikamente zielen auf eine Reaktivierung der Immunantwort
gegen den Tumor ab. Der durch einige Chemotherapeutika ausgelöste immunogene
Zelltod (ICD) macht die für das Immunsystem nicht mehr erkennbaren Tumorzellen
wieder sichtbar, da neben Antigenen auch Zytokine und Gefahrensignale freigesetzt
werden. Die Freisetzung der DAMPs kann trotz der immunmodulierenden Tumorzellen
Leukozyten wie APCs und T-Zellen anlocken und aktivieren. Dadurch werden nicht nur
die unmittelbar durch die Chemotherapie zerstörten Zellen eliminiert, sondern auch die
verbleibenden Zellklone und Mikrometastasen vom Immunsystem besser erkannt und
phagozytiert (Kepp et al. 2014, Piranlioglu et al. 2019). Der Organismus wird quasi in
situ gegen diesen Tumor geimpft (Fan und Moon 2015).
Der Goldstandard zum Nachweis der ICD-induzierenden Eigenschaften eines Medika-
ments benötigt gemäß den Konsensus-Richtlinien Impftests mit immunkompetenten, syn-
genen Mäusen, da die Untersuchung adaptiver Immunantworten ein komplexes in vivo
Immunsystem erfordert. Dafür werden zwei Ansätze vorgeschlagen: Erstens wird die
Immunität überprüft, indem Zellen, die zuvor in vitro mit dem ICD-Induktor behandelt
worden sind, einem Versuchstier als Vakzine gespritzt werden und die Immunität
anschließend mit lebenden Tumorzellen herausgefordert wird. Zweitens wird der direkte
Therapieansatz getestet, indem eine Neoplasie, die in immunkompetenten Mäusen
gewachsen ist, mittels des ICD-induzierenden Medikaments in vivo zerstört wird. Bei
erneuter Implantation derselben neoplastischen Zellreihe kann sich in der Maus kein
Tumor mehr entwickeln, da das Immunsystem des Tieres die neoplastischen Zellen
erkennt und eliminiert (Galluzzi et al. 2020).
2.6 Nebenwirkungen der systemischen Chemotherapie
Ein Hauptproblem der Chemotherapeutika, die meist intravenös appliziert werden, sind
ihre vielseitigen und schwerwiegenden Nebenwirkungen. Da vor allem schnell proli-
ferierende Zellen durch das Medikament angegriffen werden, treten Anämie und Immun-
suppression durch Schädigung des blutbildenden Systems, Mukositis und Übelkeit durch
Schädigung der gastrointestinalen Mukosa und Haarausfall häufig auf (Rossato et al.
2014). Insbesondere die Immunschwäche ist meist die dosislimitierende Nebenwirkung
(Yamamoto et al. 2019).
13Einerseits leiden die Patienten dadurch an schwerwiegenden Infektionen (Nordvig et al.
2018), andererseits arbeitet auch die Immunüberwachung des Tumors nicht mehr
zufriedenstellend. Der Hauptgrund für die weitreichenden Nebenwirkungen der
Chemotherapie ist die unkontrollierte Verteilung des löslichen Medikaments im gesamten
Körper. In Studien zur Verteilung der Zytostatika konnte gezeigt werden, dass nur ein
Bruchteil der Dosis tatsächlich den Tumor erreicht (Minchinton und Tannock 2006,
Tietze et al. 2013). So wird nicht nur die Proliferation der Tumorzellen, sondern auch
aller gesunden Zellen im Körper gehemmt, sodass insbesondere Gewebe, die sich schnell
teilen, wie das blutbildende System oder die Schleimhäute, besonders geschädigt werden.
2.7 Gezielte Krebstherapie
Mit diesem Dilemma der Nebenwirkungen durch die Therapie beschäftigt sich die
gezielte Krebstherapie (engl. targeted therapy). Durch Kopplung eines Medikaments an
verschiedene Trägerstoffe (wie beispielsweise Nanopartikel) kann der Wirkstoff am
beabsichtigten Wirkort angereichert werden. In der Nanomedizin werden meist Partikel
mit einer Größe von 1 bis 100 nm verwendet, die in ihren Eigenschaften wie Größe,
Oberflächenspannung oder Hydrophilie modelliert werden können.
Man unterscheidet den passiven Transport, der meistens durch verlängerte
Zirkulationszeiten im Blut und durch verminderten Wirkstoffabbau erreicht wird
(Lammers et al. 2012), vom aktiven Transport, bei dem der Wirkstoff an eine
tumorspezifische Komponente gekoppelt wird und sich so speziell an Tumorzellen
anreichert (Bae und Park 2011).
2.7.1 Passiver Medikamententransport
Nach intravenöser Applikation sind Nanopartikel meist auf das Gefäßsystem und auf
Organe mit gefenstertem Endothel, wie z.B. Leber und Milz, beschränkt, da die
Porengröße des normalen intakten Endothels etwa 5 nm beträgt. Tumore und entzündetes
Gewebe sind ebenfalls gut zugänglich, da sie sich durch gefenstertes Endothel und eine
Gefäßundichtigkeit (vascular leakiness) auszeichnen. Kleinere Nanopartikel werden
durch die renale Clearance mittels glomerulärer Filtration, tubuläre Sekretion und
abschließender Urinausscheidung eliminiert. Größere Partikel werden über Phagozyten
des retikuloendothelialen Systems aus dem Blutkreislauf ausgeschieden. Makrophagen in
der Leber (sogenannte Kupffer-Zellen), in der Milz und im zirkulierenden Blut nehmen
14opsonisierte Nanopartikel rasch auf und bauen sie intrazellulär ab (Gustafson et al. 2015,
Janko et al. 2019).
Für den passiven, zielgerichteten Transport werden die Wirkstoffe häufig in Liposomen
(Romeo et al. 2019, Mahner et al. 2015), Polymeren (Dinndorf et al. 2007) oder Mizellen
(Keam et al. 2019) eingebettet. Die Kopplung des Wirkstoffs an Nanopartikel bewirkt
einen verminderten Wirkstoffabbau und eine geringere Diffusion in gesundes Gewebe
mit intaktem Endothel und somit eine verlängerte Zirkulationszeit im Blut.
Die Diffusion in Tumorgewebe, das durch das schnelle Wachstum häufig insuffiziente,
löchrige Gefäßmembranen besitzt (Tanaka et al. 2004), bleibt jedoch erhalten und der an
Nanopartikel gebundene Wirkstoff reichert sich passiv im Tumorgewebe an. Hinzu
kommt, dass undifferenzierte Tumore eine unzureichende Drainage von extravasaler
Flüssigkeit durch die Lymphe aufweisen. Diese Eigenschaft von tumorösem Gewebe
wird „enhanced permeability and retention“-Effekt (EPR-Effekt) genannt (Park et al.
2018). Entscheidend sind hierfür vor allem die Größe und Oberflächenspannung der
Partikel. In soliden Tumoren mit schlecht differenzierten Kapillaren können durch den
EPR-Effekt Partikel von einer Größe bis zu 100 nm austreten.
2.7.2 Aktiver Medikamententransport
Trotz der verbesserten Akkumulation im Tumorgewebe durch den EPR-Effekt beim
passiven Medikamententransport ist der Anteil der Nanopartikel, die in den Tumor
gelangen, immer noch begrenzt. Die Mehrzahl der applizierten Nanopartikel wird
innerhalb weniger Stunden aus dem Blut entfernt und nur einige Prozent verbleiben im
Körperkreislauf (Hong et al. 2009). Daher werden beim aktiven Transport zytotoxische
Medikamente unter anderem an Antikörper gekoppelt, die gezielt Tumoroberflächen-
proteine binden (Petrylak et al. 2019, Rizzieri 2016).
Viele Tumorentitäten zeichnen sich patientenunabhängig durch typische Mutationen aus,
die zu den gleichen Antigenen und Rezeptoren translatiert werden. Diese Antigene lassen
sich nicht nur immunhistochemisch nachweisen und dienen so der Diagnostik und
Identifikation des Ursprungsgewebes, sie werden zunehmend auch als Zielstruktur für die
gezielte Krebstherapie genutzt.
Ein Beispiel ist Trastuzumab, das seit Jahren in der Therapie des Her2/neu-positiven
Mammakarzinoms zugelassen ist (Interdisziplinäre S3-Leitlinie für die Früherkennung,
Diagnostik, Therapie und Nachsorge des Mammakarzinoms). Besonders aggressive
Mammakarzinome überexprimieren den epidermalen Wachstumsfaktor-Rezeptor
15Her2/neu und befeuern so noch weiter das Tumorwachstum. Trastuzumab bindet an
Her2/neu und blockiert so gezielt die überschießende Proliferation. Es konnte gezeigt
werden, dass an Trastuzumab gebundene Wirkstoffe in Her2/neu positivem Tumor-
gewebe angereichert werden (Hathaway et al. 2011).
Auch der Folsäurerezeptor (FR) wird von vielen Tumoren, die aus dem Epiderm
stammen, überexprimiert und eignet sich daher als Zielstruktur für den aktiven Medika-
mententransport. Daher wurde eine Reihe von FR-gerichteten Wirkstoffen entwickelt,
darunter monoklonale Anti-FR-Antikörper, FR-bindende Antikörper-Wirkstoff-
Konjugate und auf Folsäure basierende niedermolekulare Wirkstoff-Konjugate (Vergote
und Leamon 2015). Es konnte gezeigt werden, dass sich mit Folsäure gekoppelte
superparamagnetische Eisenoxidnanopartikel (SPIONs) vermehrt in Folsäurerezeptor-
exprimierendem Tumorgewebe anreichern (Fan et al. 2011).
2.8 Magnetisches Drug Targeting
Als weitere Möglichkeit des aktiven Medikamententransportes ist noch die Kopplung an
magnetische Nanopartikel zu nennen, welche sich durch ein extern angelegtes Magnet-
feld anreichern lassen. Dieses Vorgehen wird als magnetische Wirkstoffanreicherung
(engl.: Magnetic drug targeting, kurz MDT) bezeichnet. Üblicherweise wird dafür das
Wirkstoffkonjugat intraarteriell in ein Tumor zuführendes Gefäß appliziert und dann
mittels eines Elektromagneten, der ein gut definiertes Magnetfeld erzeugt, für eine
bestimmte Zeitspanne am Zielort angereichert (Lyer et al. 2015). Bei den
Trägersubstanzen handelt es sich um superparamagnetische Eisenoxid-Nanopartikel
(SPIONs), deren Oberfläche mit organischen Materialien bedeckt wird, wie z.B.
Fettsäuren, Polysacchariden oder Polymeren, um die kolloidale Stabilität zu verbessern,
eine Ausfällung der Partikel aus dem Trägermedium zu verhindern (Tietze et al. 2015),
die Biokompatibilität zu erhöhen und um sie mit Wirkstoffen zu beladen. Untersucht
wurden Beschichtungen mit Hyaluronsäure (Vyas et al. 2015), Dextran (Unterweger et
al. 2018) oder Polyethylenglucol (PEG) (Pernia Leal et al. 2015).
Beispielsweise wurden SPIONs in eine Öl-Phase eingebettet, mit einer Polymerhülle und
anschließend einer PEG-Schicht ummantelt, um so magnetische Nanokapseln (m-NC)
mit einer Größe von ca. 120 nm zu bilden (Bai et al. 2016). In vivo Versuche mit CT-26-
tumortragenden Mäusen zeigten, dass das Chemotherapeutikum Docetaxel, wenn es an
m-NCs gebunden wird, mittels MDT im Tumor angereichert werden kann, und dort auch
16in gebundener Form seine immunogene, tumorverhindernde Wirkung entfaltet. So konnte
die therapeutische Wirksamkeit und das Überleben im Vergleich zur nicht zielgerichteten
Anwendung verbessert werden (Al-Jamal et al. 2016).
2.8.1 Synthese der SPIONs und Beladung mit Chemotherapeutikum
Forschungsgrundlage für die vorliegende Arbeit waren die nach der Methode von Zaloga
et al (Zaloga et al. 2016) hergestellten Eisenoxidnanopartikel. Dafür werden Eisen-II- und
Eisen-III-Salze gelöst, mit Laurinsäure beschichtet (SPIONLA) und aufgereinigt (Zaloga
et al. 2014). Dieses Ferrofluid wird mit einer sterilen Albuminlösung inkubiert. Dadurch
lagern sich die Albuminmoleküle einschichtig um die SPIONLA und bilden eine
stabilisierende Proteincorona um die Partikel (SPIONLA-HSA). Es konnte gezeigt werden,
dass diese Albuminhülle die Biokompatibilität verbessert (Zaloga et al. 2015), da
Albumin das häufigste Plasmaprotein im Blut ist. Die fertigen Partikel haben eine
hydrodynamische Größe von 58,91 nm, ein Zetapotential von -11,5 mV und eine
Eisenkonzentration von 4,87 mg/ml.
Vor jedem Experiment werden die Partikel frisch mit flüssigem MTO durch Mischen
beladen, wobei sich die MTO-Moleküle stabil in die Albuminhülle einlagern. Es entsteht
SPIONLA-HSA*MTO (im weiteren Verlauf kurz SPIONMTO genannt). Mehrere in vitro-
Settings konnten zeigen, dass sich diese Partikel mittels Magnetfeld sowohl im statischen
als auch im zirkulierenden Medium gezielt anreichern lassen und dass das gebundene
MTO sowohl in adhärenten 2D-Tumor-Modellen als auch in dreidimensionalen
Tumorsphäroiden eine Zellschädigung herbeiführt, die vergleichbar ist mit der von
ungebundenem MTO (Hornung et al. 2015, Hornung et al. 2016, Alev et al. 2018, Nguyen
et al. 2020). Gleichzeitig sind in Zellen außerhalb des Magnetfeldes keine SPIONMTO und
auch keine Schädigungen nachweisbar (Ratschker et al. 2020).
2.8.2 Magnetische Akkumulation des Wirkstoffs im Tumor
Experimente mit Plattenepithelkarzinom-tragenden Kaninchen zeigten, dass nach
Applikation von SPIONMTO in die den Tumor versorgende Arterie und nach
entsprechender Anreicherung durch ein extern angelegtes Magnetfeld über dem Tumor
die MTO-Menge in der Tumorregion von 1% nach systemischer intravenöser Applikation
auf 50-60% mit MDT i.a. erhöht werden konnte. Vollständige Tumorremissionen oder
langsameres Tumorwachstum mit verlängerten Überlebenszeiten zeigten sich bei der
Mehrzahl der behandelten Tiere (Tietze et al. 2013).
17In vivo könnte der EPR-Effekt eine zusätzliche Rolle spielen. Tumorversorgende Gefäße
werden sehr rasch gebildet, um die Energieversorgung des schnell wachsenden Tumors
zu ermöglichen. Daher sind Tumorgefäße aufgrund ihrer raschen Gefäßbildung undicht,
was eine effiziente magnetische Extravasation der Nanopartikel am Tumorort ermöglicht.
Die Nanopartikel werden magnetisch aus den Gefäßen herausgezogen. Außerdem wurde
gezeigt, dass die Partikel an der Tumorstelle bleiben und nach Entfernen des Magneten
nicht in die Blutgefäße des Kaninchens zurückwandern. Detaillierte Untersuchungen der
Biodistribution von SPIONMTO 24 Stunden nach Applikation zeigten einen deutlich
erhöhten intratumoralen Medikamentenspiegel. Dies ermöglichte die Reduzierung der
therapeutischen Dosis auf 5-10% der Dosis, die üblicherweise bei systemischer
Anwendung verwendet wurde (Tietze et al. 2013).
Umgekehrt konnte durch die Tumorakkumulation die MTO-Dosis in gesundem Gewebe
und insbesondere in peripheren Immunzellen deutlich gesenkt werden (Janko et al. 2013).
2.9 Zielgerichtete Chemotherapie mit Induktion einer Immunantwort
Die Schonung von gesunden Immunzellen ist von besonderer Bedeutung, da so die
immunogenen Effekte des MTO wieder besser zum Tragen kommen könnten. Wie schon
beschrieben, besitzt MTO die Eigenschaft, einen immunogenen Zelltod zu verursachen,
der Leukozyten, insbesondere DCs und T-Zellen, aktiviert und so auf die Antigene der
Tumorzellen aufmerksam macht. Dadurch ist das Immunsystem in der Lage, den Tumor
selbstständig zu eliminieren.
Dieser Vorgang läuft nur in immunkompetenten Patienten ab. Bei einer systemischen
Chemotherapie mit MTO kommt es allerdings immer zu einer Immunsuppression, da die
Leukozyten durch ihre physiologisch schnelle Proliferation besonders anfällig für MTO
sind. So ist MTO auch für die Therapie der Multiplen Sklerose als Immunsuppressivum
zugelassen (S2k-Leitlinie Diagnose und Therapie der Multiplen Sklerose). Der
immunogene Effekt von MTO kann somit in der Krebsbehandlung nicht oder nur
vermindert zum Therapieerfolg beitragen. Die zielgerichtete Anreicherung von
SPIONMTO mittels MDT im Tumor ermöglicht es, das Immunsystem zu schonen.
Unklar war vor Beginn des Dissertationsprojektes, ob SPIONMTO in adhärenten
Tumorzellen in gleichem Maße einen ICD induziert wie das ungebundene Medikament.
Vorhergehende Versuche mit Suspensions-Zellen (Jurkat, eine humane T-Zell-
Leukämie-Zelllinie) zeigten, dass es bezüglich der Zelltodmodalitäten von mit MTO
18behandelten Jurkat-Zellen keine Rolle spielt, ob das Medikament frei löslich oder SPION-
gebunden vorliegt (Alev et al. 2018). Da sich MDT allerdings nur bei soliden Tumoren
anwenden lässt, war ein Versuchsaufbau mit adhärenten Zellen notwendig.
Im Rahmen dieser Dissertation konnte gezeigt werden, dass es für die ICD-verursachende
Wirkung von MTO keinen Unterschied macht, ob es ungebunden oder gebunden an
SPIONMTO vorliegt. Adhärente Kolonkarzinomzellen (HT-29 Zellen) wurden bei
Behandlung sowohl mit freiem MTO als auch mit SPIONMTO apoptotisch und setzten
typische DAMPs und Tumorantigene frei. Der Überstand von sterbenden HT-29 Zellen
bewirkte eine Differenzierung von unreifen DCs zu reifen antigenpräsentierenden DCs
und erwies sich als chemotaktisch aktiv auf Monozyten, die dadurch in Richtung des
Überstandes wanderten. Eine Differenzierung von APCs und die Fähigkeit, Leukozyten
zur Migration zu bewegen, sind zwei entscheidende Schritte in der Entwicklung einer
Antitumorreaktion des Immunsystems.
In weiteren Versuchen muss nun zuerst überprüft werden, ob sich T-Zellen in vitro durch
die so differenzierten DCs primen lassen. Als nächstes wäre dann zu überprüfen, ob der
durch SPIONMTO mittels MDT verursachte ICD in vivo auch an Stellen abseits der
ursprünglichen Tumor-Lokalisation zu einer Elimination von Mikrometastasen durch das
Immunsystem führt. Ein möglicher Versuchsaufbau wäre, in Anlehnung an die
Konsensus-Richtlinien für ICD (Galluzzi et al. 2020), die in vivo Anreicherung von
SPIONMTO mittels MDT im Tumorgewebe und konsekutiver immunogener
Tumorelimination. Eine permanente Immunantwort gegen die Tumorzellen würde durch
die erneute Implantation der gleichen Tumorzellen an anderer Stelle überprüft.
Entwickelt sich nach Reimplantation der neoplastischen Zellen kein erneuter Tumor, ist
von einer erfolgreichen in vivo Immunisierung gegen den Tumor auszugehen. So kann
mit einer zielgerichteten Therapie durch SPIONMTO-Anreicherung mittels MDT der
ganze Organismus gegen den Tumor geimpft werden.
193. Publikationsliste
1. Ratschker T, Egenberger L, Alev M, Zschiesche L, Band J, Schreiber E, Frey B,
Derer A, Alexiou C, Janko C. Mitoxantrone-Loaded Nanoparticles for
Magnetically Controlled Tumor Therapy-Induction of Tumor Cell Death, Release
of Danger Signals and Activation of Immune Cells. Pharmaceutics. 2020 Sep
27;12(10):E923. doi: 10.3390/pharmaceutics12100923. PMID: 32992645.
2. Janko C, Ratschker T, Nguyen K, Zschiesche L, Tietze R, Lyer S, Alexiou C.
Functionalized Superparamagnetic Iron Oxide Nanoparticles (SPIONs) as
Platform for the Targeted Multimodal Tumor Therapy. Front Oncol. 2019 Feb
13;9:59. doi: 10.3389/fonc.2019.00059. PMID: 30815389; PMCID:
PMC6382019.
20Sie können auch lesen

















































