Praktikum Humangenetik WS 2018/19 - an der Ruhr-Universität Bochum
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Praktikum Humangenetik WS 2018/19 Ort: Praktikumsraum der Physiologie MAFO Ebene 0 Raum 222 Süd Zeit: gemäß Gruppenverteilungsplan Leitung: Dr. H.P. Nguyen, PD Dr. S. Hoffjan; Prof. Dr. B. Eiben, PD Dr. W. Klein, PD Dr. M. Meins, PD Dr. B. Miterski, Dr. W. Gerding, Dr. R. Kropatsch, Dr. G. Dekomien, Dr. M. Schlinghoff Vorbemerkung: Zur erfolgreichen Teilnahme am Praktikum ist die Kenntnis dieses Skripts unerlässlich. Dieses Skript kann jedoch kein Lehrbuch ersetzen, und deshalb sollte die Vorbereitung auch mit einem Lehrbuch erfolgen. Dies ist insbesondere zur theoretischen Aufarbeitung der angeführten Lernziele erforderlich, die vor der Praktikumsteilnahme erfolgen muss. Ablaufplan: Einleitung Die Humangenetik umfasst neben der genetischen Beratung die beiden großen Bereiche Humanzytogenetik und Molekulargenetik. Die Zytogenetik befasst sich mit Veränderungen menschlicher Chromosomen und den daraus resultierenden Fehlbildungen bzw. Entwicklungsstörungen. In der Molekulargenetik werden Verän- derungen (Mutationen) auf DNA-Sequenzebene als Ursache erblicher Erkrankungen untersucht (Abb. 1).

Molekulargenetik
Zytogenetik
Abb. 1: Bereiche der Humangenetik
Da auch in der Zytogenetik in zunehmendem Maße molekulargenetische Techniken
eingesetzt werden, um das Vorhandensein bestimmter Chromosomenabschnitte, Gene
oder DNA-Sequenzen zu überprüfen, spricht man dann von molekularer Zytogenetik. Im
Rahmen dieses Praktikums sollen Grundlagen und die wichtigsten heute verwendeten
Untersuchungstechniken für die genannten Bereiche erläutert und anhand von
Fallbeispielen verdeutlicht werden, welche Folgen für Anlageträger und deren
Angehörige sich aus den erhobenen Befunden ergeben können.
1. Grundlagen Humanzytogenetik
Der Mensch hat in allen Somazellen 2n = 46 Chromosomen, die mit Hilfe spezieller
Färbe- und Differenzierungsmethoden, den sog. Banden- oder Bänderungstechniken,
unterschieden und charakterisiert werden. Chromosomen, die mit diesen Techniken
angefärbt werden, zeigen Muster aus unterschiedlich großen dunklen, hellen oder blass
gefärbten Bereichen, den Banden (s. Abb. 2). Das Bandenmuster ist für jedes Chromo-
som spezifisch; nur homologe,
strukturgleiche Chromosomen zeigen ein
gleiches Muster.
Mit der Chromosomendiagnostik kann bei
einer Reihe von angeborenen Fehlbildun-
gen und Entwicklungsstörungen eine
chromosomale Ursache festgestellt werden.
Mit Hilfe der pränatalen Diagnostik kann
schon vor der Geburt eine Chromosomen-
analyse beim Fötus durchgeführt werden.
Abb. 2: Metaphase mit G-gebänderten menschlichen Chromosomen
1.1 Präparation von Metaphasechromosomen des Menschen
Zur routinemäßigen Darstellung von Chromosomen des Menschen sind Lymphozyten
des peripheren Bluts das am besten geeignete und am meisten verwendete Ausgangs-
material (weitere Gewebe für die Chromosomenanalyse sind in Tabelle 1 aufgeführt).
Heparinisiertes Blut wird in Kulturmedium bei 37° C drei Tage lang kultiviert. Dem
Kulturmedium wird Phytohämagglutinin (PHA) zugesetzt. PHA ist ein Mitogen und regt
T-Lymphozyten, die normalerweise im peripheren Blut keine Teilungsaktivität zeigen,
während der Kultur zu wiederholten Zellteilungen an. Etwa 30-90 min vor dem Ende der
Kultur fügt man Colcemid hinzu. Diese dem pflanzlichen Colchizin ähnliche
Verbindung unterbindet die Ausbildung des Spindelapparates der Zellen, so dass die
Chromosomen im Metaphasestadium verharren. Zu Beginn der sich dann
anschließenden Aufarbeitung werden die Zellen mit einem hypotonen Medium (0,56 %
KCl) gemischt, das ein Anschwellen der Lymphozyten und ein Auseinanderweichen der
Metaphasechromosomen bewirkt. Dadurch wird die Wahrscheinlichkeit geringer, dass
sich die Chromosomen einer Zelle beim Auftropfen von Zellsuspension auf einen
Objektträger überlagern und die Karyotypanalyse erschweren.
Tabelle 1: Gewebe, die in der Routinediagnostik eingesetzt werden
Blut postnatale Chromosomenanalyse
Hautfibroblasten Nachweis von Chromosomenaberrationen, die u.U. in
Lymphozyten nicht nachweisbar sind
Knochenmark Nachweis u.a. Leukämie-assoziierter Aberrationen
Amnionzellen; Chorionzotten pränatale Diagnostik
Abortgewebe Nachweis chromosomaler Ursachen für eine Fehlgeburt
Um das charakteristische Bandenmuster zu erhalten, müssen die Chromosomen mit
speziellen Techniken angefärbt werden. Die routinemäßig am häufigsten angewandte
Technik ist die G-Banden-Technik (auch GTG-Technik genannt nach: G-bands by Trypsin
using Giemsa). Die auf den Objektträgern aufgetropften und angetrockneten
Metaphasechromosomen werden hierzu mit Trypsin (Enzym, das Eiweiß verdaut) vor-
behandelt und dann mit einer Giemsa-Farbstofflösung gefärbt. Diese Technik erzeugt bei
Chromosomen im Metaphasestadium etwa 300-600 hell und dunkel gefärbte Banden pro
haploidem Chromosomensatz. Die chemischen Reaktionen, durch welche die
unterschiedlichen Bandentypen an den Chromosomen entstehen, sind noch nicht
vollständig geklärt. Chromosomenbereiche mit einem hohen Nicht-Histon-Protein Anteil
ergeben eher dunkle G-Banden. Offenbar enthalten diese dunklen G-Banden auch DNA-
Sequenzen mit einem relativ hohen Adenin-Thymin Gehalt, während die hellen Banden
einen hohen Guanin-Cytosin Gehalt besitzen. In den hellen G-Banden wurden auch
deutlich mehr Gene lokalisiert als in den dunklen.
1.2 Charakterisierung der menschlichen Chromosomen
Der Karyotyp des Menschen zeigt normalerweise 22 Autosomenpaare (44 Chromosomen)
und 2 Geschlechtschromosomen (Gonosomen). Die beiden Geschlechtschromosomen
der Frau sind einander homolog und werden als X-Chromosomen bezeichnet; der Mann
hat als Gonosomen ein einzelnes X-Chromosom und das für das männliche Geschlecht
charakteristische Y-Chromosom. Diese Verteilung der Geschlechtschromosomen
bewirkt, dass Söhne ihr X-Chromosom immer von der Mutter und ihr Y-Chromosom
vom Vater erhalten. Der Vater vererbt sein X-Chromosom in der Regel nur an seine
Töchter. Jedes der 46 Metaphasechromosomen des Menschen wird durch die Zentromer-
region (Spindelfaser-Ansatzstelle), die üblicherweise als primäre Einschnürung sichtbar
ist und an der die beiden Schwesterchromatiden zusammenhängen, in einen mit p
bezeichneten kurzen und in einen mit q bezeichneten langen Arm unterteilt. Die Lageder Zentromerregion ist konstant. In der Humanzytogenetik werden die Chromosomen aufgrund der Lage der Zentromerregion in drei Gruppen eingeteilt (s. Abb. 3): a) metazentrische Chromosomen: Das Zentromer liegt in der Mitte des Chromosoms oder ist der Mitte sehr nahe. b) submetazentrische Chromosomen: Das Zentromer ist deutlich von der Mitte bzw. dem Ende des Chromosoms entfernt. c) telozentrische Chromosomen: Das Zentromer befindet sich nahezu am Ende des Chromosoms. Abb. 3: Eingruppierung der Chromosomen nach Lage des Zentromers 1.3 Erstellung eines Karyogramms Um eine Chromosomenanalyse bei einem Menschen durchführen zu können, erstellt man ein Karyogramm in Form eines geordnet arrangierten Chromosomensatzes. Ein Beispiel für ein menschliches Karyogramm ist in Abb. 4 dargestellt. Abb. 4: Menschliches Karyogramm Nach internationaler Übereinkunft (Denver Convention, Paris Conference; siehe Abb. 5) werden die Chromosomen des Menschen entsprechend ihrer abnehmenden Länge von 1
bis 22 durchnummeriert. Chromosom 1 ist das größte Chromosom, Chromosom 21 das kleinste (nicht Chromosom 22, wie ursprünglich angenommen). Die Chromosomen 13, 14 und 15 sowie die kleinen Chromosomen 21 und 22 zeichnen sich gegenüber den übrigen Chromosomen jeweils durch die telozentrische Lage der Zentromerregion, durch eine Nukleolus-Organisator-Region (NOR) und einen endständigen Satelliten am kurzen Arm aus. In den NOR Regionen sind die Gene für die ribosomale RNA lokalisiert, ihre Position ist durch eine sekundäre Einschnürung markiert. Als Satelliten werden kleine, endständige (knopfähnliche) Chromosomensegmente bezeichnet, die durch die sekundäre Einschnürung vom Chromosomenkörper abgesetzt sind. Das Y-Chromosom ist ebenfalls telozentrisch, trägt aber weder Satelliten noch ribosomale Gene. Chromosomen mit einer ähnlichen Lage der Zentromerregion und ähnlicher Länge werden in Gruppen zusammengefasst. Abb. 5: Pariser Nomenklatur (Paris Conference 1971) zur Bezeichnung der Banden Bei der international gebräuchlichen, formalen Bezeichnung normaler und aberranter Karyotypen geht man folgendermaßen vor: Zuerst wird die Gesamtzahl (2n) der Chromosomen angeführt, dann folgt nach einem Komma die Geschlechtschromoso- menkonstitution. Die genaue Bezeichnung einer eventuellen Aberration steht nach den Geschlechtschromosomen geschrieben und ist von diesen durch ein weiteres Komma getrennt. So lautet die Bezeichnung für einen normalen männlichen Karyotyp 46,XY bzw. 46,XX für einen weiblichen. Ein Junge mit Down-Syndrom (freie Trisomie 21) hat die Karyotypformel 47,XY,+21. 1.4 Chromosomenstörungen Mehr als 20% aller Konzeptionen beim Menschen haben Chromosomenstörungen; durch spontane Aborte verringert sich der Anteil der Chromosomendefekte aber auf 0,6% bei Lebendgeburten. Zwei Kategorien von zytogenetisch erkennbaren Aberrationen können grundsätzlich unterschieden werden: numerische und strukturelle Chromosomenaberrationen. 1.4.1 Numerische Chromosomenaberrationen Numerische Chromosomenaberrationen zeichnen sich durch eine Veränderung der Anzahl der Chromosomen aus. Dabei können ganze Chromosomensätze zusätzlich vorhanden sein (Polyploidie) bzw. einzelne Chromosomen betroffen sein (Aneuploidie).
Die numerischen Chromosomenaberrationen des Menschen haben ihre Hauptursache in der fehlerhaften Verteilung einzelner Chromosomen bei der Keimzellenbildung der Eltern. Diese Fehlverteilungen beruhen in überwiegendem Maße auf der Nicht-Trennung (Non-disjunction) zweier homologer Chromosomen in der 1. Reifeteilung bzw. der Schwesterchromatiden eines Chromosoms in der 2. Reifeteilung. Das Resultat sind Keimzellen, denen das betreffende Chromosom fehlt und solche, die dieses Chromosom zweimal besitzen. Nach der Vereinigung mit einer normalen, haploiden Keimzelle liegt dann im ersten Fall eine Monosomie und im zweiten Fall eine Trisomie dieses Chromosoms vor. Monosomien sind beim Menschen letal, mit der Ausnahme einer Monosomie für das X-Chromosom. Neben Aneuploidien der Geschlechtschromosomen werden in der Regel nur die autosomalen Trisomien der Chromosomen 13, 18 und 21 bei lebend geborenen Kindern gefunden (Tabelle 2). Tabelle 2: Beispiele für numerische Chromosomenaberrationen: 47,XY,+21 oder 47,XX,+21 Trisomie 21 (Down-Syndrom) 47,XY,+18 oder 47,XX,+18 Trisomie 18 (Edwards-Syndrom) 47,XY,+13 oder 47,XX,+13 Trisomie 13 (Pätau-Syndrom) 45,X Turner-Syndrom 47,XXY Klinefelter-Syndrom Non-disjunction kann auch in der Mitose während der frühen Zellteilungsstadien des Keims vorkommen. Dies führt zu Karyotypen mit einem chromosomalen Mosaik. Hierbei können sowohl Zelllinien mit normalen als auch mit aberranten Chromoso- menzahlen entstehen. Mit zunehmendem Gebäralter der Mutter nimmt die Häufigkeit numerischer Chromosomenaberrationen zu. Eine Ausnahme scheint das Turner- Syndrom zu sein. Der Zusammenhang zwischen dem Gebäralter der Mutter und der Häufigkeit einer numerischen Chromosomenaberration bei den Kindern ist in Abb. 6 für die freie Trisomie 21 dargestellt. Abb. 6: Abhängigkeit des Risikos für ein Down-Syndrom vom Alter der Schwangeren. 1.4.2 Strukturelle Chromosomenaberrationen Bei den strukturellen Chromosomenaberrationen ist die Anzahl der Chromosomen in der Regel unverändert, aber bei einzelnen Chromosomen sind Veränderungen im
Chromosomenaufbau nachweisbar. Dies ist an einer Veränderung des Bandenmusters
des jeweiligen Chromosoms erkennbar. Strukturelle Aberrationen sind durch
Bruchereignisse bedingt. Man unterscheidet:
Deletionen Chromosomenstückverluste
Duplikationen Verdoppelung von Chromosomenabschnitten
Inversionen Umkehr der normalen Aufeinanderfolge von Chromosomenab--
schnitten
Insertionen Ein Stück eines Chromosoms befindet sich innerhalb eines
anderen Chromosoms bzw. Chromosomenabschnitts
Translokationen Umbau von Chromosomenstücken zwischen verschiedenen Chro-
mosomen
Zentrische Fusion Zwei telozentrische Chromosomen fusionieren zu einem
Chromosom (Robertson‘sche Translokation)
Bei Deletionen bzw. Duplikationen ist der beteiligte chromosomale Abschnitt nur einfach
bzw. dreifach vorhanden. Man bezeichnet dies als partielle Monosomie bzw. Trisomie
(partiell, weil nur ein Abschnitt eines Chromosoms und nicht das komplette Chromosom
betroffen ist). Derartige Chromosomenaberrationen sind Ursachen verschiedener
klinischer Syndrome (z. B. Katzenschrei-Syndrom [5p-], Wolf-Hirschhorn-Syndrom [4p-]).
Deletionen können so klein sein, dass sie lichtmikroskopisch nicht sichtbar sind (bei ei-
ner durchschnittlichen Bandenauflösung entspricht das einer Größe von weniger als 5-10
Megabasen). Um auch solche Veränderungen zu erfassen, kommen molekularzytogeneti-
sche Methoden wie die FISH-Hybridisierung oder Array-Untersuchung (s.u.) zum
Einsatz.
Bei Inversionen und Translokationen kann die Gesamtmenge des Erbguts unverändert
sein, d.h. es sind weder Gene verloren gegangen noch hinzugekommen. Es hat sich nur
die Abfolge der Gene verändert. Der Karyotyp wird in diesem Fall als „balanciert“
bezeichnet. So sind Träger einer balancierten Translokation bzw. Inversion häufig
symptomfrei (wenn allerdings ein Bruchpunkt innerhalb eines Gens liegt, kann daraus
eine monogen vererbte Erkrankung resultieren). Eine balancierte Translokation bei
einem Elternteil kann jedoch zu einem unbalancierten Karyotyp beim Kind führen, was
Fehlgeburten bzw. Fehlbildungen und/oder Behinderungen auslösen kann.
2. Grundlagen der Molekulargenetik
Die Molekulargenetik befasst sich mit Veränderungen auf DNA-Ebene, die im Gegensatz
zu den zytogenetischen Veränderungen nicht im Mikroskop darstellbar sind. Ein
wesentliches Ziel der molekularen Genetik ist das Verständnis für Erkrankungen, die
durch genetische Veränderungen verursacht werden, um darauf aufbauend das
diagnostische Vorgehen und therapeutische Strategien zu entwickeln bzw. zu verbessern.
Für die molekulargenetischen Analysen wird in der Regel genomische DNA verwendet,
die aus peripheren Blutleukozyten gewonnen wird; seltener kommen auch Unter-
suchungen z.B. an RNA zum Einsatz.
Die Einzelbausteine der Nukleinsäuren (DNA, RNA) sind die Nukleotide (Zucker-
Phosphat-basischer Ring, der basische Ring wird oft verkürzt auch einfach Base
genannt). Die „Wendeltreppe“ des Doppelhelix-Moleküls der DNA besteht also aus zwei
gegenläufigen Zucker-Phosphat-„Rückgraten" und den basischen Ringen als
„Treppenstufen“, die nach innen gerichtet sind. Im DNA-Doppelstrang stehen sich
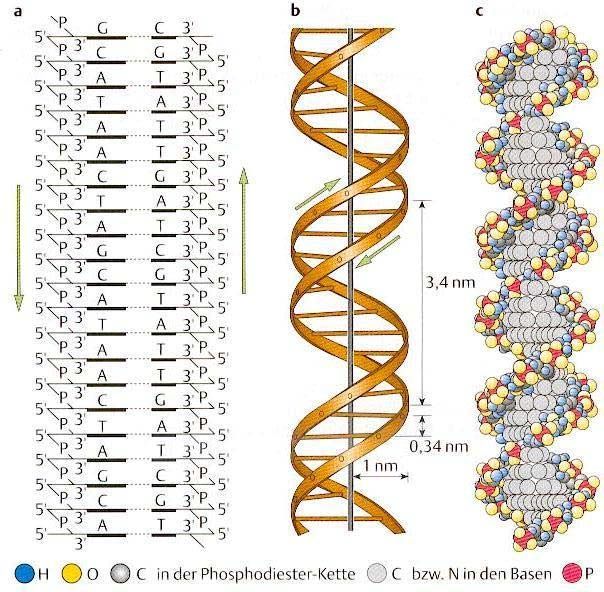
immer zwei komplementäre Basen (Abkürzung = Anfangsbuchstabe) gegenüber, welcheüber zwei (A-T) bzw. drei (G-C) stabile H-Brückenbindungen verknüpft sind (s. Abb. 7). Die Komplementarität der Nukleinsäuren wird für die meisten molekularbiologischen Untersuchungsmethoden ausgenutzt (Trennen des Doppelstrangs in die zwei Einzelstränge, Anlagern komplementärer Moleküle an Einzelstränge = Hybridisieren). Abb. 7: Struktur-Modelle der DNA-Doppelhelix; a) Die beiden ‚Zucker-Phosphat-Rückgrate’ der DNA verlaufen antiparallel; b) Dimensionen der DNA-Doppelhelix; c) Kalottenmodell. Jeder eukaryotische Organismus besitzt in jeder kernhaltigen Zelle eine bestimmte Menge an Erbgut-DNA. Sie wird Genom genannt. Die jeweiligen Genomgrößen sind charakteristisch für jede Tier- oder Pflanzenart (s. Abb. 8). Während Genome von Prokaryonten oft sehr dicht mit Erbinformationen gepackt sind (z. T. sind sogar Gene bekannt, deren Information sich überlappt), gibt es in den Genomen der meisten Eukaryonten auch große Abschnitte, die keinerlei proteinkodierende Information (keine Gene) für die Entwicklung, das Verhalten und Befinden des Organismus tragen. Abb. 8: DNA-Gehalte verschiedener Organismengruppen Das diploide Genom eines Menschen besteht aus ~7 Milliarden Nukleotidpaaren. Davon beinhalten nur ca. 1,1% kodierende Sequenzen, die tatsächlich in Protein umgeschrieben
werden, während etwa 99% nicht kodierend sind. Im 2003 veröffentlichten Humangenomprojekt wurde abgeschätzt, dass das menschliche Genom ca. 23.000 Gene enthält. Per Definition (2006) wird unter einem Gen eine „lokalisierbare Region genomischer Sequenz [verstanden], die einer Erb-Einheit entspricht, welche assoziiert ist mir regulatorischen Regionen, transkribierten Regionen und/oder anderen funktionellen Sequenzregionen“. Zur biologischen Bedeutung der DNA-Bereiche, in der keine sequenzabhängige Information enthalten ist, herrscht z.Z. noch weiterer Forschungsbedarf. Da der Austausch oder Wegfall eines Nukleotids bzw. die Umlagerung ganzer Sequenzteile innerhalb dieser Regionen meist nicht zu einer Fehlfunktion innerhalb des Organismus führt, zeichnet sich dieser Teil der DNA durch eine wesentlich größere Variabilität aus. Andererseits kann der Austausch einer einzigen Base in der kodierenden Sequenz eines Gens u.U. zum Auftreten einer schweren erblichen Erkrankung führen. Zur Untersuchung von Genen als Grundlage für bestimmte Erkrankungen kommen diverse molekulargenetische Methoden zum Einsatz. Einen hohen Stellenwert nimmt hier die Polymerasekettenreaktion (polymerase chain reaction, PCR) ein, mit der spezifische DNA-Abschnitte nahezu unbegrenzt vervielfältigt werden können und dann für weitere Analyseverfahren zur Verfügung stehen. Des Weiteren werden u.a. die DNA- Sequenzierung, bei der die Abfolge der DNA-Basen bestimmt wird, der DNA-Verdau mit Hilfe von Restriktionsenzymen und zumeist die Gelelektrophorese eingesetzt. Aufgrund der Weiterentwicklung der Technologien ist es zunehmend möglich, die Sequenzanalyse zahlreicher Gene simultan durchzuführen (sog. next generation sequencing). 3. Molekulare Zytogenetik Auch in der Zytogenetik werden in zunehmendem Maße molekulargenetische Tech- niken eingesetzt, um das Vorhandensein bestimmter Chromosomen, Gene oder DNA- Sequenzen zu überprüfen. Eine in der Humanzytogenetik häufig angewendete, diagnostische Testmethode ist die Fluoreszenz In Situ Hybridisierung (FISH). Das Prinzip dieser Methode besteht darin, eine DNA-Sequenz, die spezifisch für ein be- stimmtes Chromosom oder Gen ist, als Sonde zu verwenden und mit den Metapha- sechromosomen zu hybridisieren. Dabei muss zunächst sowohl die DNA der Sonde wie auch der Chromosomenprobe durch Denaturieren in eine einzelsträngige Form überführt werden. Dann erfolgt für die in situ Hybridisierung die Zugabe der Sonden- DNA zu den Chromosomen. Bei Vorliegen komplementärer Stränge in Sonde und Chromosom bilden sich DNA-Doppelstränge (Hybridstränge) aus Sonden- und Proben- DNA. Zum Zweck der Markierung wird die Sonden-DNA mit fluoreszierenden Sub- stanzen versehen. Im Fluoreszenzmikroskop fluoreszieren dann diejenigen Chromo- somen oder chromosomale Regionen, an welche die Sonde hybridisiert hat. Die FISH Methode erlaubt es, auch an Interphasekernen nachzuprüfen, ob ein Karyotyp mit einer numerischen Chromosomenaberration vorliegt oder welche DNA-Sequenzen in Tu- morgeweben amplifiziert wurden. Voraussetzung ist wieder die Verfügbarkeit einer chromosomen- bzw. tumorspezifischen Sonden-DNA. Bei Trisomie 21 können z.B. unter Verwendung einer Chromosom 21 spezifischen DNA-Sequenz als Sonde in den meisten Interphasekernen drei fluoreszierende Bereiche nachgewiesen werden. Eine weitere zunehmend eingesetzte Methode stellt die Chip-basierte Array-Analyse dar, die zum Nachweis bzw. Ausschluss unbalancierter chromosomaler Aberrationen (Deletionen oder Zugewinne) dient, die zu klein sind, um im Lichtmikroskop gesehen zu werden. Das Auflösungsvermögen ist dabei um ein Vielfaches höher als bei der konventionellen Karyotypisierung.
4. Lernziele
Zytogenetik:
•Die unterschiedlichen Chromosomenaberrationen sollen aufgezählt werden und
das Prinzip ihrer Entstehung erklärt werden können.
• Die beim Menschen vorkommenden numerischen Abberationen, die mit dem Le-
ben vereinbar sind, sollen benannt und deren wichtigste Symptome beschrieben
werden können.
• Beispiele von Krankheitsbildern, die durch strukturelle Abberationen verursacht
werden, sollten bekannt sein.
• Die Folgen struktureller Aberrationen für Anlageträger und deren Angehörige
sollen erklärt werden können.
Molekulargenetik:
• Der Aufbau der DNA und verschiedene Mutationen auf DNA-Ebene sollen erklärt
werden können.
• Das Prinzip der Gelelektrophorese und ihre praktische Umsetzung sollen
vermittelt werden.
• Es soll ein Einblick in moderne molekulargenetische Techniken gegeben werden
können.
• Die Untersuchung und Bedeutung der Faktor V-Leiden Mutation soll erläutert
werden können.
5. Versuchsmaterialien und Durchführung der Agarose-Gelelektrophorese
Hintergrund: Risiko für Thromboembolie bei Mutation im Faktor V-Gen (Faktor V-
Leiden Mutation)
Thrombosen kommen bei ~1:1000 Personen pro Jahr vor. Die Faktor V-Leiden Mutation
ist der häufigste genetisch bedingte Defekt, der mit venösen thromboembolischen
Erkrankungen assoziiert ist. Faktor V ist an der Gerinnungs-Kaskade beteiligt und kann
physiologischerweise durch aktiviertes Protein C (über proteolytische Spaltung des
Faktors Va) inaktiviert werden. Die „Leiden“ Mutation im Faktor V-Gen ist durch einen
Nukleotid-Austausch verursacht, hierdurch kommt es zum Aminosäure-Austausch.
Durch diese Veränderung wird die hemmende Aktivität des Proteins C auf den Faktor V
herabgesetzt (aktivierte Protein C-Resistenz) und somit das Gleichgewicht zugunsten
gerinnungsfördernder Reaktionen verschoben.
Etwa 5 % der westlichen Bevölkerung sind heterozygote Träger eines Faktor V-Leiden
Allels, bei diesen Personen ist das individuelle Thromboserisiko bis 10-fach erhöht. Für
homozygote Träger der Faktor V-Leiden Mutation ist das Risiko für Thrombosen sogar
bis 100-fach erhöht. Orale Kontrazeptiva erhöhen zusätzlich das Thromboserisiko für
mutations-betroffene Frauen. Eine Kombination der Faktor V-Leiden Mutation mit
anderen genetisch bedingten Störungen (z.B. Prothrombin-Polymorphismen) kann das
individuelle Thromboserisiko weiter steigern.
Bei folgenden Indikationen ist es auf Wunsch der Risikoperson sinnvoll, auf Faktor V-
Leiden Mutation zu testen: Personen mit erhöhtem Thromboserisiko (positive Familienanamnese für
Thromboembolien, zur Abklärung rezidivierender Thrombosen)
Patientinnen mit thromboembolischen Komplikationen während der
Schwangerschaft oder unter Einnahme oraler Kontrazeptiva.
Für Untersuchungen auf die Leiden-Mutation im Faktor V Gen wird eine Blutprobe
(EDTA-antikoaguliert) benötigt, aus der DNA extrahiert wird.
Im Rahmen des Praktikums werden zur Verfügung gestellte PCR-Produkte des Faktor V-
Gens im elektrischen Feld mittels der Agarose-Gelelektrophorese entsprechend der
Fragmentlänge nach Restriktionsverdau aufgetrennt. Diese Technik stellt eine essentielle
Methode des molekularbiologischen Labors dar, die dort tagtäglich vielfach angewendet
wird.
Mittels PCR wird der Bereich des fraglichen Nukleotid-Austauschs im Faktor V Gen
vervielfältigt und mit einem Restriktionsenzym gespalten. Der Verdau findet nur statt,
wenn das mutierte Allel M vorhanden ist, nicht im Normalfall (N) ohne Mutation.
Abbildung 9 zeigt die unterschiedlichen Fragmentgrößen bei einer Kontrollperson (1),
einem heterozygoten (2) und einem homozygoten (3) Träger der Faktor V-Leiden
Mutation nach Auftrennung durch Gelelektrophorese.
Abb. 9: Agarose-Gelabbildung mit drei verschiedenen DNA Fragmentmustern
Versuchsmaterialien:
o Feinwaage, (Mini-)Gelkammern mit Gieß-Kämmen, Spannungsquelle (power supply)
mit Kabeln, UV-Leuchtschirm (cave: Hornhaut-und Linsenepithel-Schädigungen,
daher sind Schutzvorrichtungen nötig: Plexiglasplatte, Schutzbrille)
o Agarose und Gelelektrophoresepuffer (Herstellung: siehe chemisches Praktikum)
o SERVA DNA Stain (Farbstoff zur Detektion von Nukleinsäuren)
o DNA-Lösungen
o Längenmarker
o Gel-Ladepuffer incl. Ladefarbe (Ficoll, Bromphenolblau, Xylencyanol)Durchführung:
1. Feinwaage: Abwiegen des Agarose-Pulvers für 50 ml eines 1,5-2%igen Gels
2. Agarose suspendieren, lösen (aufkochen); nach Zugabe von SERVA DNA Stain
auf ~50°C abkühlen lassen
3. Vorbereiten der Kammer (Abdichten, Geltaschen-Kamm einsetzen)
4. Flüssige Agarose-Lösung in Gelkammer gießen (Gel verfestigt sich nach
Abkühlen)
5. Gel mit Elektrophorese-Puffer überschichten (1-2 mm über Geloberfläche)
6. Gel-Kamm entfernen
7. Geltaschen mit mit Ladepuffer versetzten DNA-Lösungen und Längenmarker be-
laden
8. Spannung (60-100 Volt) anlegen für ~20 min
9. Spannungsgeber abschalten, Kabel entfernen, Transport zum UV-Leuchtschirm;
UV-Belichtung des Gels (cave: UV-Licht), Analyse des Gels: Banden und DNA-
Fragmentlängenbestimmung durch Vergleich mit Längenmarkerbanden (pUC19-
Plasmid-DNA verdaut mit Restriktionsenzym: 2686 Basenpaare (bp), 501 bp, 489
bp, 404 bp, 331 bp, 242bp, 190 bp, 147 bp, 111 bp, 110 bp, 67 bp, 34 bp, 34 bp,
26bp).
Abb. 10: Vorbereiten, Gießen, Erstarren lassen und Beladen eines horizontalen Agarosegels mit DNA-
ProbenHintergrundinformation: Isolieren, Schneiden (= Verdauen = Spalten = Restringieren)
und Auftrennen der DNA
Zur DNA -Isolierung eignen sich alle Gewebe oder Zellen, die Zellkerne enthalten. Dabei
ist es gleichgültig, ob sie aus Blut, Sperma, Urin, Speichel, Haarwurzeln oder einem
beliebigen Gewebe stammen. Die DNA wird nach den für die einzelnen Zelltypen
optimierten Verfahren isoliert. Es ist möglich, isolierte DNA mittels eines DNA-
spaltenden Enzyms (Restriktionsendonuklease) zu verdauen. Diese Enzyme erkennen
bestimmte DNA-Sequenzen und durchtrennen den DNA-Doppelstrang an dieser Stelle.
So erkennt z.B. die Restriktionsendonuklease Hae III die Sequenz
.….G G C C…..
…..C C G G…..
und schneidet sie, wie mit dem Pfeil dargestellt. Durch diesen Verdau erhält man DNA-
Fragmente unterschiedlicher Länge. Diese Fragmente können mittels Gelelektrophorese
im elektrischen Feld aufgetrennt werden. Dabei macht man sich den Umstand zunutze,
dass DNA-Fragmente unterschiedlicher Länge verschieden schnell durch ein Gel
wandern: Die DNA-Fragmente sind aufgrund ihrer molekularen Struktur (Zucker-
Phosphat-Rückgrat) negativ geladen. Legt man elektrische Spannung an ein Gel, das man
sich am besten als ein engmaschiges dreidimensionales Gitternetz vorstellt, so wandern
die einzelnen Fragmente wegen ihrer negativen Ladung in dem elektrischen Feld in
Richtung Anode (+ Pol). Lange Fragmente bewegen sich aufgrund ihrer Größe
langsamer durch das engmaschige Gitter als kürzere. DNA-Fragmente lassen sich also
auf diese Weise entsprechend ihrer Länge auftrennen. Die Gele befinden sich hierzu
meist in wässrigen Puffer-(Elektrolyt-) Systemen. Mit derartigen Methoden werden
Längenunterschiede der DNA (und RNA) darstellbar. Unterschiedliche Medien haben
unterschiedliche Trennbereiche (Standard-Agarosegele 50-25000 Basen, verschiedene
Polyacrylamidgele von 1-1500 Basen). Die Darstellung der DNA-Fragmente im Gel erfolgt
durch Beigabe des Farbstoff SERVA DNA Stain, welcher bei der Bindung an
Nukleinsäuren grüne Fluoreszenz emittiert.
Verschieden lange DNA-Fragmente werden gelelektrophoretisch gemäß ihrem
Molekulargewicht aufgetrennt: kurze Fragmente laufen schneller durch die Gelmatrix,
lange DNA-Moleküle langsamer. Mittels sequenzspezifischer Sonden werden aus der
Vielzahl der möglichen nur einzelne DNA-Fragmente an ihrer charakteristischen
Wanderungsposition im Gel dargestellt.
Literatur
Hirsch-Kauffmann, M. und M. Schweiger (2009): Biologie für Mediziner und
Naturwissenschaftler (7. Auflage). Georg Thieme Verlag StuttgartSie können auch lesen

















































