Beam-Search zum automatisierten Entwurf und Scoring neuer ROR-Liganden mithilfe maschineller Intelligenz - Research Collection
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
ETH Library Beam-Search zum automatisierten Entwurf und Scoring neuer ROR- Liganden mithilfe maschineller Intelligenz Journal Article Author(s): Moret, Michael; Helmstädter, Moritz; Grisoni, Francesca ; Schneider, Gisbert ; Merk, Daniel Publication date: 2021-08-23 Permanent link: https://doi.org/10.3929/ethz-b-000501729 Rights / license: Creative Commons Attribution 4.0 International Originally published in: Angewandte Chemie. International Edition 133(35), https://doi.org/10.1002/ange.202104405 Funding acknowledgement: 182176 - De novo molecular design by deep learning (SNF) This page was generated automatically upon download from the ETH Zurich Research Collection. For more information, please consult the Terms of use.
Angewandte
Forschungsartikel Chemie
Zitierweise: Angew. Chem. Int. Ed. 2021, 60, 19477 – 19482
De-novo-Design Internationale Ausgabe: doi.org/10.1002/anie.202104405
Deutsche Ausgabe: doi.org/10.1002/ange.202104405
Beam-Search zum automatisierten Entwurf und Scoring neuer ROR-
Liganden mithilfe maschineller Intelligenz**
Michael Moret+, Moritz Helmst-dter+, Francesca Grisoni, Gisbert Schneider* und Daniel Merk*
Abstract: Chemische Sprachmodelle ermçglichen ein De-no-
vo-Wirkstoff-Design ohne explizite chemische Konstruktions-
regeln. W-hrend solche Modelle angewendet wurden, um
neuartige Verbindungen mit angestrebter biologischer Aktivit-t
zu generieren, bleibt die tats-chliche Priorisierung und Aus-
wahl der vielversprechendsten Molekglentwgrfe („Designs“)
eine Herausforderung. Wir haben hier die von chemischen
Sprachmodellen gelernten Wahrscheinlichkeiten mithilfe des
Beam-Search-Algorithmus als Modell-intrinsische Technik fgr
das Molekgldesign und die Bewertung der Designs („Sco-
ring“) genutzt. Die prospektive Anwendung dieser Methode
fghrte zu neuartigen inversen Agonisten der Retinoid-related-
Orphan-Rezeptoren (RORs). Jedes Design war in drei Reak-
tionsschritten synthetisierbar und zeigte eine niedrig-mikro-
molare bis nanomolare Potenz gegengber RORg. Als Modell-
intrinsische Technik eliminiert das Beam-Search-Sampling die
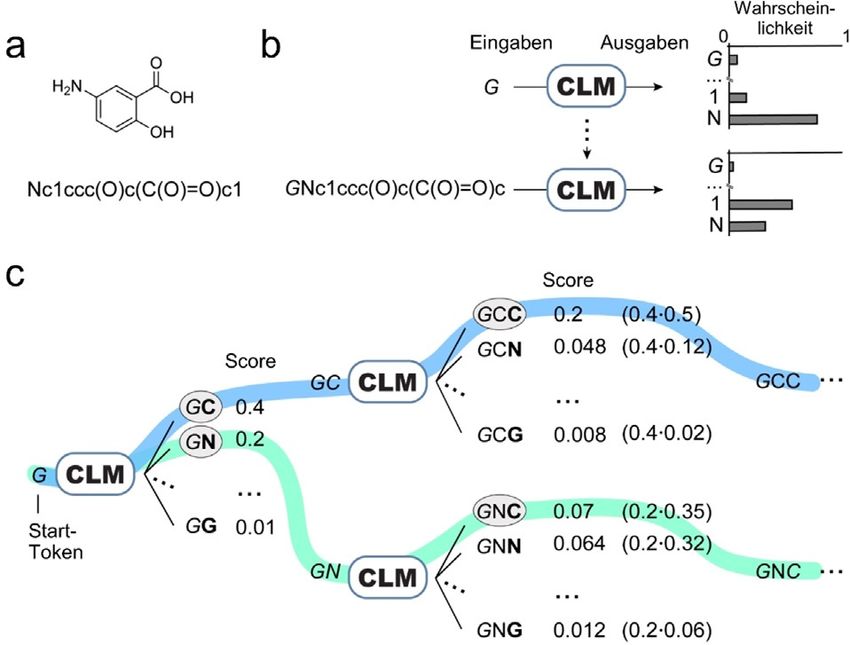
strikte Notwendigkeit externer Molekgl-Scoring-Funktionen Abbildung 1. Molekfldesign durch chemische Sprachmodelle (CLM)
und erweitert damit die Anwendbarkeit generativer kgnstlicher und Beam-Search-Sampling. a) Kekul8-Struktur eines Beispielmolekfls
Intelligenz in der datengetriebenen Wirkstoffforschung. mit seinem korrespondierenden SMILES-String. b) CLM-Training. Das
CLM lernt die Wahrscheinlichkeit jedes SMILES-Zeichens („Token“) ba-
sierend auf den vorherigen Token im SMILES-String vorherzusagen.
Einleitung
c) Beam-Search mit k = 2: Der Algorithmus beh-lt die zwei wahr-
scheinlichsten SMILES-Strings im Blick (farblich hervorgehoben). In
Generatives Deep Learning,[1, 2] eine Klasse maschineller diesem Beispiel erfolgt die Generierung des SMILES-Strings von links
Lernmodelle, die in der Lage sind, neue Daten zu generieren, nach rechts.
kann fgr das computergestgtzte De-novo-Design pharmako-
logisch aktiver Verbindungen eingesetzt werden.[3–5] Deep-
Learning-basierte Algorithmen fgr das Molekgldesign kçn- mçglicht.[12] Frghere Studien haben gezeigt, dass chemische
nen spezifische chemische Merkmale aus „rohen“ Molekgl- Sprachmodelle (Chemical Language Models, CLMs),[13, 14]
darstellungen, wie z. B. molekularen Graphen und dem Sim- insbesondere auf SMILES-Strings trainierte generative
plified Molecular Input Line Entry System (SMILES, Ab- Deep-Learning-Modelle, neuartige Molekgle mit experi-
bildung 1 a)[11] extrahieren,[6–10] was ihnen potenziell den Zu- mentell validierter biologischer Aktivit-t generieren kçn-
gang zu unerforschten Regionen des chemischen Raums er- nen.[9, 15, 16] CLMs haben dabei die F-higkeit bewiesen, fo-
[*] M. Moret,[+] Prof. Dr. F. Grisoni, Prof. Dr. G. Schneider Prof. Dr. D. Merk
ETH Zurich, Department of Chemistry and Applied Biosciences LMU Mfnchen, Department of Pharmacy
Vladimir-Prelog-Weg 4, 8093 Zurich (Schweiz) Butenandtstraße 7, 81377 Mfnchen (Deutschland)
E-Mail: gisbert@ethz.ch [+] Diese Autoren haben zu gleichen Teilen zu der Arbeit beigetragen.
M. Helmst-dter,[+] Prof. Dr. D. Merk [**] Eine frfhere Version dieses Manuskripts ist auf einem Preprint-
Goethe University Frankfurt Server hinterlegt worden (http://doi.org/10.26434/chemrxiv.
Institute of Pharmaceutical Chemistry 14153408.v1).
Max-von-Laue-Straße 9, 60438 Frankfurt (Deutschland)
Hintergrundinformationen und Identifikationsnummern (ORCIDs)
E-Mail: merk@pharmchem.uni-frankfurt.de
der Autoren sind unter:
Prof. Dr. F. Grisoni https://doi.org/10.1002/ange.202104405 zu finden.
Eindhoven University of Technology
T 2021 Die Autoren. Angewandte Chemie verçffentlicht von Wiley-
Institute for Complex Molecular Systems
VCH GmbH. Dieser Open Access Beitrag steht unter den Be-
Department of Biomedical Engineering
dingungen der Creative Commons Attribution License, die jede
Groene Loper 7, 5612AZ Eindhoven (Niederlande)
Nutzung des Beitrages in allen Medien gestattet, sofern der ur-
Prof. Dr. G. Schneider sprfngliche Beitrag ordnungsgem-ß zitiert wird.
ETH Singapore SEC Ltd
1 CREATE Way, #06-01 CREATE Tower
Singapore 138602 (Singapur)
19626 T 2021 Die Autoren. Angewandte Chemie verçffentlicht von Wiley-VCH GmbH Angew. Chem. 2021, 133, 19626 – 19632Angewandte
Forschungsartikel Chemie
kussierte chemische Merkmale aus kleinen Sammlungen von Orphan-Rezeptor (ROR)-Liganden[26] wurde das Beam-Se-
Template-Molekglen mittels Transferlernen zu erler- arch-Sampling und -Scoring erfolgreich validiert.
nen.[15, 17, 18] Die Methode des Transferlernens ermçglicht die Die RORs wurden als molekulare Zielstrukturen ausge-
Wiederverwendung von zuvor gelerntem Wissen in einer w-hlt, da sie attraktive, aber nicht umfassend untersuchte
neuen Aufgabe, fgr die nur wenige Daten verfggbar sind, und potenzielle Wirkstofftargets darstellen. Sie bilden eine Fa-
wird in zwei Schritten durchgefghrt. Im ersten Schritt wird ein milie Ligand-aktivierter Transkriptionsfaktoren, die haupt-
Modell mit einer großen Menge von Daten trainiert, die sich s-chlich als Monomere agieren und unter anderem an der
auf die zu erfgllende Aufgabe beziehen („Pre-Training“). Im zirkadianen Kontrolle der Energiehomçostase[27, 28] und der
Falle von CLMs wird dies gblicherweise mit großen Mole- Regulation des Immunsystems[29, 30] beteiligt sind. RORs be-
kglsammlungen in der Grçßenordnung von 200 000 bis sitzen vielversprechendes pharmakologisches Potenzial in
1 000 000 Molekglen erreicht.[9, 16, 17] Das Pre-Training ermçg- verschiedenen Indikationen, insbesondere bei Autoimmun-
licht es dem generativen Modell, a) die SMILES-Syntax (d. h. erkrankungen.[29, 30] Bis heute hat jedoch noch kein ROR-Li-
wie alphanumerische Zeichen zusammengesetzt werden gand eine Arzneistoffzulassung erreicht, was zum Teil auf
sollten, um Strings zu generieren, die validen Molekglen substanzspezifische Schwachpunkte wie schlechte Wasser-
entsprechen, Abbildung 1) und b) die Eigenschaften des Pre- lçslichkeit, mangelnde Selektivit-t und klinische Sicher-
Trainingsdatensatzes, wie z. B. die physikochemischen Ei- heitsbedenken zurgckzufghren ist.[29, 31, 32]
genschaften und die chemische Synthetisierbarkeit der Mo-
lekgle im Datensatz zu erfassen. Im zweiten Schritt wird das
vortrainierte CLM mit einer kleineren Anzahl aufgabenspe- Ergebnisse und Diskussion
zifischer Molekgle weiter trainiert („Fine-Tuning“).[13, 19, 20]
W-hrend dieses Transferlernprozesses wird das CLM auf den Chemisches Sprachmodell und Beam-Search-Sampling zum De-
chemischen Raum von Interesse ausgerichtet, also auf Mo- novo-Design
lekgle mit den angestrebten biologischen und physikoche-
mischen Eigenschaften. Diese F-higkeit, von wenigen Daten Als mçgliche Alternative zum Temperatur-Sampling in
zu lernen („few-shot learning“[21, 22]), macht CLMs besonders Kombination mit einer externen Priorisierungs-Methode ha-
fgr die Anwendung auf biologische Zielstrukturen wertvoll, ben wir den Beam-Search-Algorithmus[33] zur Erzeugung von
fgr die nur wenige Liganden bekannt sind. Das vollst-ndig Molekglen aus einem CLM untersucht. Basierend auf den
trainierte CLM kann dann verwendet werden, um neue Mo- Wahrscheinlichkeiten, die ein CLM erlernt, kann theoretisch
lekgle in Form von SMILES-Strings zu entwerfen. Diese eine große Anzahl von SMILES-Strings erzeugt werden, es ist
Datengenerierung erfolgt durch die schrittweise Vorhersage rechnerisch aber nicht machbar, alle mçglichen Strings zu
von jeweils einem Zeichen („Token“) eines SMILES-Strings generieren. Es kann jedoch die Hypothese aufgestellt werden,
basierend auf allen vorherigen Token. Erw-hnenswert ist dass die Wahrscheinlichkeit fgr die Erzeugung eines be-
dabei, dass dieser Prozess keine vordefinierten Regeln fgr das stimmten SMILES-Strings mit der Qualit-t des korrespon-
Molekgldesign erfordert, da CLMs nur von den SMILES- dierenden Molekgls im Hinblick auf das Design-Ziel korre-
Strings lernen, die fgr das Training verwendet werden. liert, wie es im Fine-Tuning-Set repr-sentiert ist (z. B. ange-
Bisherige Anwendungen von CLMs im De-novo-Design strebte biologische Aktivit-t, physikochemische Eigenschaf-
haben das sog. Temperatur-Sampling genutzt, um große vir- ten). Mithilfe einer heuristischen Methode wie der Beam-
tuelle Molekglbibliotheken zu generieren.[9, 13, 15] Temperatur- Search kçnnen die wahrscheinlichsten Strings, die ein CLM
Sampling erlaubt es, neue SMILES-Strings zu erstellen, in- generieren kann, gefunden werden.
dem Token zum (wachsenden) String entsprechend den vom W-hrend der Molekglgenerierung durch den Beam-Se-
CLM gelernten Wahrscheinlichkeiten hinzugefggt werden, arch-Algorithmus („Beam-Search-Sampling“) fggt der Al-
wobei wahrscheinlichere Token an einer bestimmten Position gorithmus schrittweise Token zu einem SMILES-String hinzu,
h-ufiger gew-hlt werden (Abbildung 1 b). Allerdings sind die w-hrend er die k wahrscheinlichsten SMILES-Strings beh-lt.
so generierten SMILES-Strings ggf. nicht immer chemisch Um ein neues Token hinzuzufggen, berechnet der Algorith-
sinnvoll (invalide Strings), oder sie stimmen aufgrund der mus die bedingte Wahrscheinlichkeit jedes mçglichen Token
Zufallskomponente des Temperatur-Samplings nicht mit der basierend auf den Token im bestehenden String und definiert
Merkmalsverteilung der Trainingsdaten gberein. Daher be- die k wahrscheinlichsten Token, um den String zu erweitern
darf es gblicherweise zus-tzlicher Methoden, um die viel- (Abbildung 1 c). Die Menge der k wahrscheinlichsten ge-
versprechendsten Designs aus den virtuellen Molekglbiblio- w-hlten Optionen basiert auf einer Bewertungsfunktion
theken auszuw-hlen. Dies geschieht z. B. basierend auf der („Beam-Search-Score“), die als Produkt der Wahrschein-
ihnlichkeit zu bekannten biologisch aktiven Molekglen, lichkeiten der einzelnen Token berechnet wird (Abbil-
mittels externer Aktivit-tsvorhersage oder durch sog. Be- dung 1 c). Dieser Prozess wird so lange wiederholt, bis der
lohnungsfunktionen.[9, 13, 15, 23] SMILES-String vollst-ndig ist (d. h. das „Ende-des-Strings“-
Als Modell-intrinsische Alternative zum Temperatur- Token hinzugefggt wird) oder eine vordefinierte maximale
Sampling nutzen wir hier den Beam-Search-Algorithmus, der String-L-nge erreicht ist. Auf diese Weise kann der Beam-
dem CLM die gleichzeitige Erstellung und Priorisierung der Search-Algorithmus verwendet werden, um basierend auf
molekularen Designs in automatisierter Weise ohne zus-tz- 1) dem zugrundeliegenden Modell und 2) dem Beam-Search-
liche Selektionsmethoden ermçglicht.[24, 25] In einer prospek- Score hochwahrscheinliche SMILES-Strings zu erzeugen.
tiven Anwendung zur Entwicklung neuer Retinoid-related- Der Beam-Search-Score erlaubt es dabei, diese De-novo-
Angew. Chem. 2021, 133, 19626 – 19632 T 2021 Die Autoren. Angewandte Chemie verçffentlicht von Wiley-VCH GmbH www.angewandte.de 19627Angewandte
Forschungsartikel Chemie
Designs nach der Wahrscheinlichkeit ihrer SMILES-Strings Designs zu erhalten, die drei Eigenschaften besitzen: 1) eine
zu ordnen. von Naturstoffen inspirierte chemische Struktur, 2) chemi-
Zur Untersuchung und Anwendung des Beam-Search- sche Synthetisierbarkeit und 3) biologische Aktivit-t an
Sampling haben wir ein kgrzlich verçffentlichtes CLM ge- RORg. Um alle drei Ziele w-hrend des Transfer-Lernens zu
nutzt, das auf einem rekurrenten neuronalen Netzwerk mit erfgllen, wurde das zuvor mit biologisch aktiven Molekglen
sog. long short-term memory cells (LSTM) basiert, welche fgr aus ChEMBL[17] vortrainierte CLM mithilfe eines syntheti-
Sequenzmodellierung geeignet sind.[34] Das CLM wurde mit schen RORg-Liganden und vier in der Literatur[30] beschrie-
den SMILES-Strings von 365 063 Molekglen aus ChEMBL[35] benen RORg-modulierenden Naturstoffen verfeinert (Ab-
trainiert, iterativ das n-chste Token jedes SMILES-Strings bildung S1). Von diesem CLM wurde ab der fgnften Epoche
unter Bergcksichtigung der vorhergehenden Token vorher- des Fine-Tunings mit dem Beam-Search-Sampling begonnen,
zusagen (Abbildung 1 b). Die Trainingsprozedur wurde gber um sicherzustellen, dass das CLM die molekularen Merkmale
zehn Epochen durchgefghrt, was bedeutet, dass jedes fgr das des kleinen Fine-Tuning-Datensatzes ausreichend erfasst
Training verwendete Molekgl vom CLM zehnmal gesehen hatte.
wurde. Durch Transfer-Lernen („Fine-Tuning“) mit Sets be- Alle ggltigen SMILES-Strings, die das CLM zwischen den
kannter ROR-Liganden (Abbildung S1, Tabelle S1) wurde Epochen 5 und 16 (letzte Epoche des Fine-Tunings) gene-
dann in das vortrainierte CLM ein Bias in Richtung des De- rierte, wurden anhand des Beam-Search-Scores eingestuft.
sign-Ziels, n-mlich die Entwicklung neuer Molekgle mit Die fgnf Designs mit dem hçchsten Beam-Search-Score
biologischer Aktivit-t an den RORs, eingefghrt. Die Open- (Abbildung 2 a) wurden jedoch von Medizinalchemikern als
Source-Codes fgr das CLM und den Beam-Search-Algorith- synthetisch unzug-nglich eingestuft, und auch die Vorhersa-
mus sowie die in dieser Studie verwendeten Daten sind unter gen eines maschinellen Lernalgorithmus fgr retrosynthetische
https://github.com/ETHmodlab/ Analysen (IBM RXN)[40] konnten fgr keines dieser Molekgle
molecular design with beam search verfggbar. eine Syntheseroute finden. W-hrend das CLM also die
ihnlichkeit zu Naturstoffen erfasste, erfgllte es nicht das
generische Designkriterium der Synthetisierbarkeit. Diese
Anwendung des Beam-Search-Samplings zum Design inverser Ergebnisse deuten einen Nutzen des Beam-Search-Samplings
RORg-Agonisten an, die wahrscheinlichsten Designs eines CLMs zu offenbaren
und den Erfolg des Fine-Tunings hinsichtlich der Design-
In einer prospektiven Analyse wurde der Beam-Search- Ziele zu bewerten.
Algorithmus auf das Design Naturstoff-inspirierter RORg- Um diese Ergebnisse zu verbessern, wurde ein zweites
Liganden angewendet. Als traditionelle Inspirationsquelle fgr Experiment mit einer zweistufigen Fine-Tuning-Strategie
die Arzneimittelentwicklung[36, 37] kann das Lernen von Nat- durchgefghrt, bei dem das vortrainierte CLM zun-chst fgr
urstoffen gegengber rein synthetischen Molekglen mehrere 20 Epochen mit 255 synthetischen RORg-Liganden aus dem
Vorteile bieten. Naturstoffe weisen insgesamt mehr struktu- US-Patent-Subset der Protein Data Bank[41] (255 Molekgle,
relle Vielfalt, grçßere Dreidimensionalit-t und h-ufig eine Tabelle S1) trainiert wurde, um sowohl biologische Aktivit-t
hçhere Selektivit-t auf.[38, 39] Daher strebten wir an, De-novo- als auch Synthetisierbarkeit zu erfassen. Anschließend wurde
Abbildung 2. Hçchstklassifizierte, durch Beam-Search-Sampling erhaltene Designs. a) Einfaches Fine-Tuning, b) zweistufiges Fine-Tuning. R-nge
basieren auf dem Beam-Search-Score der Designs. Die hhnlichkeitswerte der hçchstklassifizierten Designs aus dem zweistufigen Fine-Tuning
Experiment beziehen sich auf die auf Morgan-Fingerprints (L-nge = 1024, Radius = 2 Bindungen) berechnete Tanimoto-hhnlichkeit zum -hnlichs-
ten bekannten aktiven Molekfl mit einem IC50-Wert an RORg in ChEMBL (Strukturen sind in Abbildung S2 gezeigt).
19628 www.angewandte.de T 2021 Die Autoren. Angewandte Chemie verçffentlicht von Wiley-VCH GmbH Angew. Chem. 2021, 133, 19626 – 19632Angewandte
Forschungsartikel Chemie
das CLM mit vier RORg-modulierenden Naturstoffen[30] teten also darauf hin, dass das zweistufige Fine-Tuning-Ver-
(Abbildung S1) fgr 16 Epochen mit dem Ziel weitertrainiert, fahren die Designziele erfgllte, sodass der zweistufige Ansatz
das Modell in Richtung Naturstoff-ihnlichkeit zu beeinflus- fgr die prospektive Anwendung gew-hlt wurde.
sen. Wie im ersten Experiment wurden dann alle ggltigen Der Vergleich der Beam-Search Designs aus dem zwei-
SMILES-Strings untersucht, die das CLM durch Beam-Se- stufigen Fine-Tuning-Verfahren mit den Trainingsmolekglen
arch-Sampling zwischen den Epochen 5 und 16 des (zweiten) und mit bekannten RORg-Modulatoren (Abbildung 3 a,b)
Fine-Tuning-Schrittes generierte. Die fgnf in diesem zweiten zeigte, dass das Beam-Search-Sampling die Erkundung des
Ansatz designten Molekgle mit dem hçchsten Beam-Search- chemischen Raums jenseits jener Regionen erlaubte, die von
Score (Abbildung 2 b) waren gem-ß IBM RXN[40] synthetisch den Molekglen des Fine-Tunings besetzt sind, obwohl dieses
zug-nglich; fgr jedes Design wurde eine Syntheseroute ge- Sampling-Verfahren die wahrscheinlichsten Token bei der
funden. Die computergenerierten Molekgle besaßen außer- Erzeugung neuer SMILES-Strings beggnstigt und nur eine
dem Naturstoffcharakteristika (Abbildung 3, Tabelle S2), was begrenzte Zahl an Mçglichkeiten untersucht. Im Vergleich zu
sich durch einen hohen Anteil an sp3-hybridisierten Kohlen- den in ChEMBL annotierten inversen RORg-Agonisten (IC50
stoffatomen (Fsp3) ausdrgckte. Die Top-5-Designs wiesen < 1 mm) waren die Beam-Search-Designs außerdem struktu-
Fsp3-Werte zwischen 50 % und 75 % auf, was mit den Werten rell vielf-ltiger im Hinblick auf die durch Morgan-Finger-
fgr die MEGx-Naturstoffbibliothek (Analyticon Discovery prints[42] dargestellten Substrukturfragmente (Abbildung 3 b).
GmbH, rel. 09-01-2018) vergleichbar war und die Fsp3-Werte Gleichzeitig besaßen die Designs aber hinsichtlich ihrer
der fgr das Pre-Training verwendeten ChEMBL-Molekgle dreidimensionalen Gestalt und Partialladungsverteilung
(51 : 30 % bzw. 33 : 20 %) gberstieg. Diese Ergebnisse deu- (dargestellt durch die „Weighted Holistic Atom Localization
and Entity Shape [WHALES]“-Deskriptoren[43, 44]) ihnlich-
keit zu den bekannten aktiven Molekglen. Offenbar lernte
das CLM also zus-tzlich zur SMILES-Syntax auch gewisse
„semantische“ Strukturmerkmale, die fgr die Bindung an
Makromolekgle relevant sind, wie z. B. molekulare Form und
Partialladungsmuster.
Prospektive experimentelle Validierung
Auf Grundlage der Beam-Search-Scores wurden drei
Designs zur Synthese und In-vitro-Charakterisierung ausge-
w-hlt. Von den fgnf Designs mit hçchstem Beam-Search-
Score (Abbildung 2 b) w-hlten wir Molekgle 1 und 2 vom
ersten und dritten Rang aus. Verbindung 2 zeigte dabei die
hçchste Tanimoto-ihnlichkeit (Morgan-Fingerprints) zu ei-
nem bekannten RORg-Modulator (Abbildung 2 b). Die
Grundgergste der Verbindungen 1 und 2 waren auch unter
Beam-Search-Designs jenseits der Top 5 verbreitet, was auf
strukturelle Pr-ferenzen hindeutete. Das „Scaffold“ (Mole-
kglgergst) von 1 fand sich im sechstplatzierten Design wieder,
und die Molekgle auf den R-ngen 10 und 13 wiesen eine hohe
ihnlichkeit zu Verbindung 2 auf, weshalb zus-tzlich Ver-
bindung 3 dieses prominenten Chemotyps von Rang 13 fgr
eine prospektive Validierung ausgew-hlt wurde. Die Verbin-
dungen 1–3 wurden gem-ß Schema 1 synthetisiert.
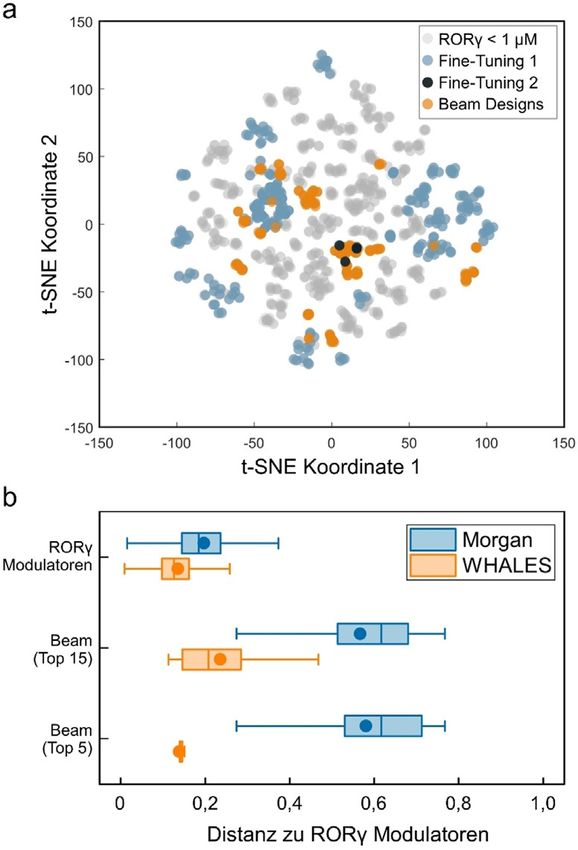
Abbildung 3. Charakteristika der CLM-Designs nach doppeltem Fine- Zur Herstellung von 1 wurden zun-chst (4-Chlorphe-
Tuning. a) t-Verteilte stochastische Nachbareinbettung (t-SNE)[45] der nyl)piperazin (4) und 4-Brombutylacetat (5) durch nukleo-
Molekflsets gem-ß Morgan-Fragment-Fingerprints (L-nge = 1024, Ra- phile Substitution zu 6 umgesetzt. Nach alkalischer Hydrolyse
dius = 2 Bindungen, Tanimoto-hhnlichkeit). Die beiden Fine-Tuning-
der Esterschutzgruppe in 6 wurde aus dem freien Alkohol 7
Sets, die in ChEMBL enthaltenen RORg-Liganden (IC50 < 1 mm,
1091 Molekfle) und die Beam-Search-Designs sind gezeigt. b) Ver- durch Mitsunobu-Reaktion mit 8-Azaspiro[4.5]decan-7,9-
gleich der Beam-Search-Designs mit bekannten RORg-Liganden dion (8) das Design 1 erhalten. Die Synthese von Design 2
(IC50 < 1 mm) hinsichtlich Morgan-Fragment-Fingerprints („Morgan“) begann ausgehend von 4-Brom-2-fluorbenzaldehyd (9), wel-
sowie dreidimensionaler Gestalt und Partialladungsverteilung („WHA- cher durch reduktive Aminierung mit Cyclobutanamin (10)
LES“). Die paarweise Distanzverteilung zwischen in ChEMBL enthalte- das sekund-re Amin 11 lieferte. Das Zwischenprodukt 11
nen RORg-Liganden ist zum Vergleich gezeigt. Ffr Morgan-Finger- wurde anschießend mit dem Sulfonylchlorid 12 zu 13 umge-
prints ist die Tanimoto-Distanz gezeigt, ffr WHALES die skalierte eu-
setzt, bevor 13 im letzten Reaktionsschritt unter Buchwald-
klidische Distanz. „Beam (Top 15)“ und „Beam (Top 5)“ beziehen sich
auf die 5 bzw. 15 hçchstklassifizierten Beam-Search-Designs. Die Hartwig-Bedingungen mit 14 das Design 2 ergab. Das struk-
Boxplots zeigen die 25., 50. und 75. Perzentile (Linien), Mittelwerte turell verwandte Design 3 wurde, ausgehend von einer nu-
(Kreis) und Ausreißergrenzen (Whisker). kleophilen aromatischen Substitution mit 4-Trif-
Angew. Chem. 2021, 133, 19626 – 19632 T 2021 Die Autoren. Angewandte Chemie verçffentlicht von Wiley-VCH GmbH www.angewandte.de 19629Angewandte
Forschungsartikel Chemie
Schema 1. Synthese der CLM-Designs 1, 2 und 3. Reagenzien und Bedingungen: a) DMF, 4-DMAP, 60 8C, 16 h, 48 %; b) KOH, H2O/THF/MeOH,
MW, 100 8C, 30 min, 98 %; c) DIAD, PPh3, THF, 0 8C!RT, 16 h, 42 %; d) NaB(OAc)3H, HOAc, DCE, RT, 50 h, 73 %; e) 4-DMAP, Pyridin, CH2Cl2,
Reflux, 16 h, 37 %; f) Pd2(dba)3, Xantphos, Cs2CO3, 1,4-Dioxan, Reflux, 16 h, 18 %; g) K2CO3, DMSO, Reflux, 48 h, 82 %.
luormethylpiperidin (14) und 4-Fluorbenzaldehyd (15), gber
Tabelle 1: Biologische Aktivit-t der CLM-Designs 1, 2 und 3 an den RORs
eine alternative Route synthetisiert. Die nukleophile aroma- in Gal4-Hybrid-Reportergen-Assays. Daten sind als Mittelwert : S.E.M.
tische Substitution lieferte eine hçhere Ausbeute (siehe dargestellt, n + 4.
Schema 1) als die Buchwald-Hartwig-Reaktion, konnte aber
IC50 [mm]
wegen der mçglichen Bildung von Regioisomeren nicht fgr Struktur und ID RORa RORb RORg
die Synthese von 2 angewendet werden. Reduktive Aminie-
rung des Substitutionsproduktes 16 mit Cyclobutanamin (10)
zu 17, gefolgt von einer Sulfonamidbildung mit Phenyl-
methansulfonylchlorid (12), lieferte Design 3. > 10 > 10 4,6 : 0,5
Die In-vitro-Charakterisierung der Verbindungen 1, 2 und
3 in Gal4-ROR-Hybrid-Reportergen-Assays best-tigte den
angestrebten inversen RORg-Agonismus mit mikromolaren
bis submikromolaren IC50-Werten (Tabelle 1). Die gem-ß
23 : 3 22 : 1 0,37 : 0,05
Beam-Search-Score ranghçchste Verbindung 1 wirkte der
basalen RORg-Aktivit-t mit einem IC50-Wert von 4,6 mm
entgegen. Sie war auch an RORa und RORb aktiv, genaue
IC50-Werte konnten aber aufgrund von Zytotoxizit-t nicht
bestimmt werden. Die Verbindungen 2 und 3 zeigten inversen 10 : 1 7,6 : 0,5 0,68 : 0,07
RORg-Agonismus mit IC50-Werten von 0,37 mm (2) bzw.
0,68 mm (3). Neben dem angestrebten inversen RORg-Ago-
nismus wiesen alle drei synthetisierten Designs eine ausge-
pr-gte Pr-ferenz fgr den RORg-Subtyp auf, wobei die Ver-
bindungen 2 und 3 im Vergleich zu den verwandten RORa- Fazit
und RORb-Isoformen eine mehr als zehnfach hçhere Potenz
an RORg besaßen. Diese Ergebnisse zeigen, dass das CLM Der Beam-Search-Algorithmus wurde zum De-novo-
mit Beam-Search-Sampling die biologische Aktivit-t der Design mit einem CLM angewendet. Dabei erzeugte und
Trainingsmolekgle in den De-novo-Designs konservierte. bewertete der Algorithmus die Designs automatisch, ohne
dass zus-tzliche Priorisierungsregeln erforderlich waren. Die
19630 www.angewandte.de T 2021 Die Autoren. Angewandte Chemie verçffentlicht von Wiley-VCH GmbH Angew. Chem. 2021, 133, 19626 – 19632Angewandte
Forschungsartikel Chemie
prospektive experimentelle Validierung der Methode lieferte
neue Molekgle, die mit inversem Agonismus am Ligand-ak- [1] J. Schmidhuber, Neural Networks 2015, 61, 85 – 117.
[2] Y. Lecun, Y. Bengio, G. Hinton, Nature 2015, 521, 436 – 444.
tivierten Transkriptionsfaktor RORg die angestrebte biolo-
[3] H. Chen, O. Engkvist, Y. Wang, M. Olivecrona, T. Blaschke,
gische Aktivit-t besaßen und unterschiedliche Grade an
Drug Discov. Today 2018, 23, 1241 – 1250.
ihnlichkeit zu bekannten RORg-Modulatoren aufwiesen [4] W. P. Walters, R. Barzilay, Acc. Chem. Res. 2021, 54, 263 – 270.
(0,28–0,71 Tanimoto-ihnlichkeit auf Morgan-Fingerprints). [5] B. Sanchez-Lengeling, A. Aspuru-Guzik, Science 2018, 361,
Der Beam-Search-Algorithmus in Verbindung mit einem 360 – 365.
CLM bewahrte also offenbar strukturelle Merkmale, die fgr [6] R. Glmez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hern#n-
die angestrebte biologische Aktivit-t notwendig sind, entwarf dez-Lobato, B. S#nchez-Lengeling, D. Sheberla, J. Aguilera-
dabei aber strukturell diverse Molekgle. Dieses Ergebnis Iparraguirre, T. D. Hirzel, R. P. Adams, A. Aspuru-Guzik, ACS
best-tigt Beam-Search-Sampling als eine geeignete Technik Cent. Sci. 2018, 4, 268 – 276.
[7] N. de Cao, T. Kipf, arXiv 2018, https://arxiv.org/abs/1805.11973.
zum De-novo-Design bioaktiver Molekgle durch ein CLM.
[8] A. Gupta, A. T. Mgller, B. J. H. Huisman, J. A. Fuchs, P.
Die rechnerischen und experimentellen Ergebnisse weisen Schneider, G. Schneider, Mol. Inf. 2018, 37, 1700111.
außerdem auf zwei attraktive Eigenschaften des Beam-Se- [9] D. Merk, L. Friedrich, F. Grisoni, G. Schneider, Mol. Inf. 2018,
arch-Algorithmus in dieser Anwendung hin. Zum einen zeigt 37, 1700153.
der Beam-Search-Algorithmus durch die Suche nach den [10] J. Bradshaw, B. Paige, M. J. Kusner, M. H. S. Segler, J. M.
wahrscheinlichsten Molekglen, die ein CLM generieren kann, Hern#ndez-Lobato, arXiv 2020, https://arxiv.org/abs/2012.11522.
die Eignung des Modells fgr die zu untersuchende Aufga- [11] D. Weininger, J. Chem. Inf. Comput. Sci. 1988, 28, 31 – 36.
benstellung auf. Die Auswertung der Beam-Search Designs [12] M. A. Skinnider, R. G. Stacey, D. S. Wishart, L. J. Foster,
ChemRxiv 2021, https://doi.org/10.26434/CHEMRXIV.
erlaubt die 3berprgfung, ob die Molekglentwgrfe mit den
13638347.V1.
Designzielen gbereinstimmen, und damit eine Bewertung des [13] M. H. S. Segler, T. Kogej, C. Tyrchan, M. P. Waller, ACS Cent.
Fine-Tuning-Erfolgs. Dies steht im Gegensatz zum her- Sci. 2018, 4, 120 – 131.
kçmmlichen Temperatur-Sampling, das Chemiker dazu ver- [14] W. Yuan, D. Jiang, D. K. Nambiar, L. P. Liew, M. P. Hay, J.
leiten kçnnte, Designs in Betracht zu ziehen, die gem-ß dem Bloomstein, P. Lu, B. Turner, Q. T. Le, R. Tibshirani, P. Khatri,
Modell nicht wahrscheinlich sind. Zum anderen kçnnte der M. G. Moloney, A. C. Koong, J. Chem. Inf. Model. 2017, 57, 875 –
Beam-Search-Score, der eine intrinsische Klassifizierung er- 882.
mçglicht, die Notwendigkeit der externen Priorisierung von [15] D. Merk, F. Grisoni, L. Friedrich, G. Schneider, Commun. Chem.
2018, 1, 68.
Designs gberwinden. Es ist jedoch zu beachten, dass die
[16] Y. Yang, R. Zhang, Z. Li, L. Mei, S. Wan, H. Ding, Z. Chen, J.
Anzahl der Designs, die durch Beam-Search erstellt werden
Xing, H. Feng, J. Han, H. Jiang, M. Zheng, C. Luo, B. Zhou, J.
kçnnen, begrenzt ist, w-hrend Temperatur-Sampling eine Med. Chem. 2020, 63, 1337 – 1360.
praktisch unendliche Anzahl von chemischen Strukturen ge- [17] M. Moret, L. Friedrich, F. Grisoni, D. Merk, G. Schneider, Nat.
nerieren kann. Beide Techniken erg-nzen sich gegenseitig Mach. Intell. 2020, 2, 171 – 180.
und bieten jeweils Vorteile. Die angestrebte Anwendung [18] M. Awale, F. Sirockin, N. Stiefl, J. L. Reymond, J. Chem. Inf.
sollte die Wahl der einen oder anderen Strategie leiten. Wenn Model. 2019, 59, 1347 – 1356.
zukgnftige prospektive Studien diese Beobachtungen best-- [19] J. Yosinski, J. Clune, Y. Bengio, H. Lipson, Adv. Neural Inf.
tigen, kçnnte das Beam-Search-Sampling dazu beitragen, die Process. Syst. 2014, 27, 3320 – 3328.
[20] M. Peters, S. Ruder, N. A. Smith, arXiv 2019, https://arxiv.org/
Anwendbarkeit von CLMs fgr das De-novo-Design in der
abs/1903.05987.
medizinischen Chemie zu st-rken. [21] H. Altae-Tran, B. Ramsundar, A. S. Pappu, V. Pande, ACS Cent.
Sci. 2017, 3, 283 – 293.
[22] Y. Wang, Q. Yao, J. Kwok, L. M. Ni, arXiv 2019, https://arxiv.org/
Acknowledgements abs/1904.05046.
[23] X. Yang, J. Zhang, K. Yoshizoe, K. Terayama, K. Tsuda, Sci.
Diese Forschung wurde durch den Schweizer Nationalfonds Technol. Adv. Mater. 2017, 18, 972 – 976.
(grant no. 205321_182176 an G.S.), die RETHINK Initiative [24] P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C.
Bekas, A. A. Lee, ACS Cent. Sci. 2019, 5, 1572 – 1583.
der ETH Zgrich und die Novartis Forschungsstiftung (Free-
[25] D. Grechishnikova, Sci. Rep. 2021, 11, 321.
Novation grant „AI in Drug Discovery“ an G.S.) gefçrdert.
[26] G. Benoit, A. Cooney, V. Giguere, H. Ingraham, M. Lazar, G.
Open Access Verçffentlichung ermçglicht und organisiert Muscat, T. Perlmann, J. P. Renaud, J. Schwabe, F. Sladek, M. J.
durch Projekt DEAL. Tsai, V. Laudet, Pharmacol. Rev. 2006, 58, 798 – 836.
[27] D. P. Marciano, M. R. Chang, C. A. Corzo, D. Goswami, V. Q.
Lam, B. D. Pascal, P. R. Griffin, Cell Metab. 2014, 19, 193 – 208.
Interessenkonflikt [28] Y. Hoon Kim, M. A. Lazar, Endocr. Rev. 2020, 41, 707 – 732.
[29] V. B. Pandya, S. Kumar, Sachchidanand, R. Sharma, R. C. Desai,
G.S. erkl-rt einen mçglichen finanziellen Interessenkonflikt J. Med. Chem. 2018, 61, 10976 – 10995.
[30] L. A. Solt, T. P. Burris, Trends Endocrinol. Metab. 2012, 23, 619 –
als Grgnder der inSili.com GmbH, Zgrich, und in seiner Rolle
627.
als Berater der pharmazeutischen Industrie. [31] S. Asimus, R. Palm8r, M. Albayaty, H. Forsman, C. Lundin, M.
Olsson, R. Pehrson, J. Mo, M. Russell, S. Carlert, D. Close, D.
Stichwçrter: De-novo-Design · Deep Learning · Kernrezeptor · Keeling, Br. J. Clin. Pharmacol. 2020, 86, 1398 – 1405.
Neuronale Netze · Wirkstoffforschung [32] D. J. Kojetin, T. P. Burris, Nat. Rev. Drug Discovery 2014, 13,
197 – 216.
Angew. Chem. 2021, 133, 19626 – 19632 T 2021 Die Autoren. Angewandte Chemie verçffentlicht von Wiley-VCH GmbH www.angewandte.de 19631Angewandte
Forschungsartikel Chemie
[33] B. T. Lowerre, PhD Thesis, Carnegie Mellon Univ. Pittsburgh, [41] H. M. Berman, T. Battistuz, T. N. Bhat, W. F. Bluhm, P. E.
1976. Bourne, K. Burkhardt, Z. Feng, G. L. Gilliland, L. Iype, S. Jain, P.
[34] S. Hochreiter, J. Schmidhuber, Neural Comput. 1997, 9, 1735 – Fagan, J. Marvin, D. Padilla, V. Ravichandran, B. Schneider, N.
1780. Thanki, H. Weissig, J. D. Westbrook, C. Zardecki, Acta Cry-
[35] A. P. Bento, A. Gaulton, A. Hersey, L. J. Bellis, J. Chambers, M. stallogr. Sect. D 2002, 58, 899 – 907.
Davies, F. A. Krgger, Y. Light, L. Mak, S. McGlinchey, M. No- [42] H. L. Morgan, J. Chem. Doc. 1965, 5, 107 – 113.
wotka, G. Papadatos, R. Santos, J. P. Overington, Nucleic Acids [43] F. Grisoni, D. Merk, V. Consonni, J. A. Hiss, S. G. Tagliabue, R.
Res. 2014, 42, D1083 – D1090. Todeschini, G. Schneider, Commun. Chem. 2018, 1, 44.
[36] D. J. Newman, G. M. Cragg, J. Nat. Prod. 2020, 83, 770 – 803. [44] F. Grisoni, G. Schneider, Methods Mol. Biol. 2021, 2266, 11 – 35.
[37] T. Rodrigues, D. Reker, P. Schneider, G. Schneider, Nat. Chem. [45] L. van der Maaten, G. Hinton, J. Mach. Learn. Res. 2008, 9,
2016, 8, 531 – 541. 2579 – 2605.
[38] P. Ertl, A. Schuffenhauer, in Prog. Drug Res., Birkh-user, Basel,
2008, S. 217 – 235.
[39] P. Ertl, S. Roggo, A. Schuffenhauer, J. Chem. Inf. Model. 2008,
48, 68 – 74. Manuskript erhalten: 30. M-rz 2021
[40] P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeusel- Ver-nderte Fassung erhalten: 2. Juni 2021
mann, R. Pisoni, C. Bekas, A. Iuliano, T. Laino, Chem. Sci. 2020, Akzeptierte Fassung online: 24. Juni 2021
11, 3316 – 3325. Endggltige Fassung online: 19. Juli 2021
19632 www.angewandte.de T 2021 Die Autoren. Angewandte Chemie verçffentlicht von Wiley-VCH GmbH Angew. Chem. 2021, 133, 19626 – 19632Sie können auch lesen