EINFÜHRUNG IN PLANUNG UND DURCHFÜHRUNG KLINISCHER PRÜFUNGEN - PD DR. MED. THOMAS SUDHOP - BFARM
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

Einführung in Planung und
Durchführung Klinischer
Prüfungen
PD Dr. med. Thomas Sudhop
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 1
I.
Inhaltsverzeichnis
II.
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 2
Arzneimittelenwicklung
Präklinisches Zellen
Entwicklungs-
programm
Tiermodelle
Phase I
Klinisches Phase II
Entwicklungs-
programm
Phase III
Phase IV
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 3
Risiken & Chancen der
Arzneimittelentwicklung
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 4
Phase I
„Human Pharmacology“
• Erste Anwendung am Menschen
– Normalerweise Probanden
– Ev. Patienten (z.B. Zytostatika)
• Sicherheit und Verträglichkeit
• Pharmakokinetik und Pharmakodynamik
• Metabolisierung und Interaktionen
• Erste Dosis-/Wirkungsbeziehungen, wenn möglich
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 5
Phase II
„Therapeutic Exploratory“
• Untersuchung zur Indikation
– symptomtragenden Probanden
– Patienten
Wirkung
• Dosis-/Wirkungsbeziehungen
• Dosisabschätzung für Folgestudien Dosis
• Datenerhebung zur Planung von Bestätigungsstudien
– Design
– Endpunkte
– Methodik
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 6
Phase III
„Therapeutic Confirmatory“
• Wirksamkeitsnachweis
– Beweis der Effizienz
• Sicherheits- und Verträglichkeitsprofil
• Datengrundlage zur Abschätzung der Nutzen/Risikoabwägung zur
Zulassung
Zulassung bei EMA, BfArM oder PEI
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 7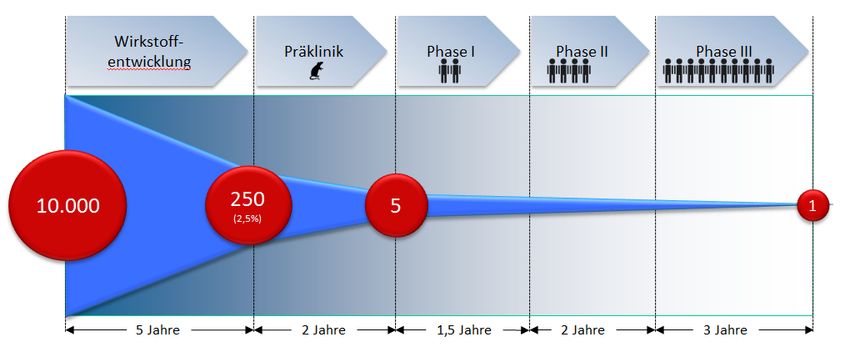
Phase IV und IIIb
• Phase IV: Klinische Prüfungen innerhalb der Zulassungsbedingungen
– Größere Populationen, besondere Populationen
– Seltene Nebenwirkungen
– Vergleichende Studien („Head-to-Head Studien“)
• Phase IIIb: Klinische Prüfungen außerhalb der Zulassungsbedingungen
– Neue Populationen
– Neue Indikationen
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 8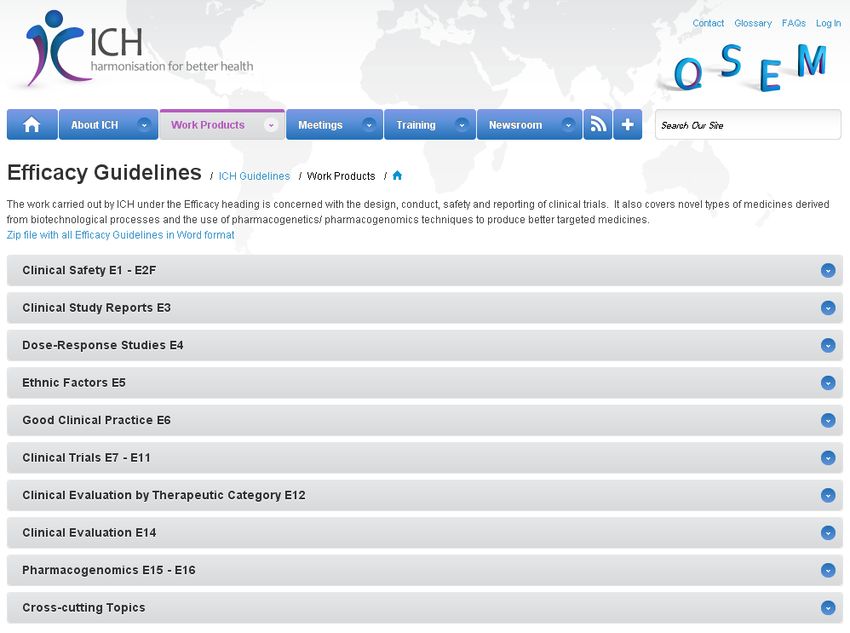
Rechtliche
Grundlagen
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 9
Leitlinie Deklaration von Helsinki (1964, 2008, 2013*) 1964+
ICH Topic E 6
„Note for Guidance on Good Clinical
Leitlinie 1995
Practice“
CPMP/ICH/135/95 „alias“ ICH GCP
Richtlinie 2001/20/EG
des EUROPÄISCHEN PARLAMENTS UND DES RATES
EU-Richtlinie vom 4. April 2001 … über die Anwendung der
2001
(Gesetzesgrundlage) Guten Klinischen Praxis (GCP) bei der Durchführung
von
klinischen Prüfungen mit Humanarzneimitteln
Arzneimittelgesetz Rechtsverordnung
Gesetz (AMG, 12. Novelle) nach §42 Abs. 3 AMG 2004
GCP-Verordnung
(GCP-V)
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 10http://www.gesetze-im-internet.de/amg_1976/
Relevante AMG Bereiche
• § 4 Abs. AMG
– Definitionen: Abs. 14 (Herstellen), 23 (Prüfung), 24 (Sponsor), 25 (Prüfer)
• § 40 AMG
– Allgemeine Voraussetzungen der klinischen Prüfung
• § 41 AMG
– Besondere Voraussetzungen der klinischen Prüfung (Patienten)
• § 42 AMG
6. Abschnitt
– Verfahren bei der Ethik-Kommission, Genehmigungsverfahren bei der Bundesoberbehörde,
Ermächtigungsgrundlage für die GCP-Verordnung
• 42a AMG
– Rücknahme und Widerruf
• 42b AMG
– Ergebnisberichte Klinischer Prüfungen
• § 67 Abs. 1 und 3 AMG
– Anzeigepflichten gegenüber Behörden
• §§ 96 und 97 AMG
– Straf- und Bußgeldvorschriften
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 11GCP-Verordnung
• „Verordnung über die Anwendung der Guten Klinischen Praxis bei der
Durchführung von klinischen Prüfungen mit Arzneimitteln zur
Anwendung am Menschen“
• Beinhaltet nicht die „Gute Klinische Praxis“ (GCP) selbst
– GCP ist in der ICH E6 Leitlinie definiert
• Regelt Definitionen, Kennzeichnung von Prüfpräparaten (IMPs),
Antragstellung und Meldepflichten in klinischen Prüfungen
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 12GCP-Verordnung (GCP-V)
Abschnitt 1 - Allgemeine Vorschriften Abschnitt 4 - Dokumentations- und
§ 1 Zweck der Verordnung
Mitteilungspflichten, Datenbanken,
§ 2 Anwendungsbereich
§ 3 Begriffsbestimmungen
Inspektionen
§ 12 Anzeige-, Dokumentations- und
Abschnitt 2 - Anforderungen an Prüfpräparate Mitteilungspflichten des Prüfers
§ 4 Herstellung und Einfuhr
§ 13 Dokumentations- und
§ 5 Kennzeichnung von Prüfpräparaten
§ 6 Entblindung in Notfallsituationen und Mitteilungspflichten des Sponsors
Rücknahme § 14 Mitteilungspflichten der
zuständigen Bundesoberbehörde
Abschnitt 3 - Genehmigung durch die
Bundesoberbehörde und Bewertung durch die § 15 Inspektionen
Ethik-Kommission
§ 7 Antragstellung
§ 8 Bewertung durch die Ethik-Kommission Abschnitt 5 - Übergangs- und Schlussbestim-
§ 9 Genehmigung durch die zuständige mungen
Bundesoberbehörde
§ 10 Nachträgliche Änderungen
§ 16 Ordnungswidrigkeiten
§ 11 Maßnahmen zum Schutz vor § 17 Übergangsbestimmungen
unmittelbarer Gefahr
§ 18 Inkrafttreten
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 13Guidelines und Bekanntmachungen
• ICH-Guidelines
– International Conference on Harmonization
– ICH Guideline zu GCP (ICH-GCP)
• Internationale Ethische Guidelines
– Deklaration von Helsinki
• EU-Guidelines
– Teilweise identisch zu ICH-Guidelines
– CT Guidance-Texte
• CT-1 zur Antragstellung bei den Behörden
• CT-2 zur Antragstellung bei den Ethik-Kommissionen
• CT-3 zur Meldung von Nebenwirkungen aus klinischen Prüfungen
• …
– Eudralex-Volume 10: Zusammenfassung aller regulatorischen Texte zu klinischen Prüfungen der EU
• Nationale Bekanntmachungen der BOB und des BMG
– 3. Bekanntmachung zur Klinischen Prüfung
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 14GXP – „Gute … Praxis“
• Leitlinien zu Qualitätsansprüchen im Rahmen der
Arzneimittelentwicklung und -herstellung
– GCP: Good Clinical Practise
– GLP: Good Laboratory Practise
– GMP: Good Manufacturing Practice
– …
• Harmonisierung dieser Guidelines über den ICH-Prozess
– ICH: International Conference on Harmonization
– Harmonisierung in den Regionen Nordamerika, Japan, EU
– Beteiligte Einrichtungen: Industrieverbände und Behörden
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 15Großes Bild innerhalb
ICH: International Conference on
des Satzspiegels
Harmonisation
Gegründet von 6 Parteien aus 3 Regionen
• EU Kommission
• European Federation of Pharmaceutical Industries and
Associations (EFPIA)
• Japan Ministry of Health, Labour and Welfare
• Japan Pharmaceutical Manufacturers Association
(JPMA)
• Food and Drug Administration (FDA)
• Pharmaceutical Research and Manufacturers of
America (PhRMA) PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 16© 2013 ICH (ICH.org)
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 17PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 18
Wichtige ICH Guidelines
• ICH E6 - Guideline for Good Clinical
Practice
• ICH M3 - Non-clinical safety studies
for conduct of human clinical trials
• ICH S6 - Preclinical safety evaluation
of biotechnology-derived
• ICH S8 - Immunotoxicity Studies for
Human Pharmaceuticals
• ICH S9 - Nonclinical Evaluation for
Anticancer Pharmaceuticals
• ICH Q Diverse Qualitäts-
Guidelines
http://www.ich.org/products/guidelines.html
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 19Gute Klinische Praxis (GCP)
Die gute klinische Praxis umfasst einen Katalog international anerkannter
ethischer und wissenschaftlicher Qualitätsanforderungen, die bei
– der Planung,
– Durchführung und
– Aufzeichnung klinischer Prüfungen an Menschen
– sowie der Berichterstattung über diese Prüfungen
eingehalten werden müssen.
Die Einhaltung dieser Praxis gewährleistet, dass die Rechte, die Sicherheit und
das Wohlergehen der Teilnehmer an klinischen Prüfungen geschützt werden
und dass die Ergebnisse der klinischen Prüfungen glaubwürdig sind.
Artikel 2 der EU-Richtlinie 2001/20
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 20Klinische Prüfung
Definition § 4 Abs. 23 AMG
Klinische Prüfung bei Menschen ist jede am Menschen durchgeführte
Untersuchung, die dazu bestimmt ist,
PD – klinische oder pharmakologische Wirkungen von Arzneimitteln zu
erforschen oder nachzuweisen oder
ADR – Nebenwirkungen festzustellen oder
PK – die Resorption, die Verteilung, den Stoffwechsel oder die Ausscheidung zu
untersuchen,
mit dem Ziel, sich von
– der Unbedenklichkeit oder
– Wirksamkeit der Arzneimittel zu überzeugen.
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 21Voraussetzungen zur Durchführung
einer klinischen Prüfung
• Vorhandensein eines Sponsors mit Sitz in der EU (ggf. gesetzlichen
Vertreter)
• Leitung durch einen verantwortlichen Prüfer
• Abschluss einer Versicherung nach § 40 AMG (Ausnahme u.U. Phase IV
Studien)
• Zustimmende Bewertung durch zuständige Ethik-Kommission (EK)
• Genehmigung durch zuständige Bundesoberbehörde (BOB)
• Anzeige bei den zuständigen Behörden (=Landesbehörden)
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 22Sponsor
Sponsor ist eine natürliche oder juristische Person, die die Verantwortung
für die Veranlassung, Organisation und Finanzierung einer klinischen
Prüfung bei Menschen übernimmt
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 23Prüfer
• Prüfer ist der für die Durchführung der klinischen Prüfung in einer
Prüfstelle verantwortliche Arzt (z.B. Praxis-Arzt)
• Wird in einer Prüfstelle die klinische Prüfung von mehreren Personen
durchgeführt, ist der verantwortliche Leiter dieser Gruppe der Prüfer
(z.B. Chefarzt, Oberarzt)
• Wird eine klinische Prüfung in mehreren Prüfstellen durchgeführt,
ernennt der Sponsor einen Prüfer zum Leiter der klinischen Prüfung
[LKP]
§ 4 Abs. 25 AMG
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 24Prüfer
• Prüfer ist der für die Durchführung der klinischen Prüfung in einer
Prüfstelle verantwortliche Arzt (z.B. Praxis-Arzt)
• Wird in einer Prüfstelle die klinische Prüfung von mehreren Personen
durchgeführt, ist der verantwortliche Leiter dieser Gruppe der Prüfer
(z.B. Chefarzt, Oberarzt)
• Wird eine klinische Prüfung in mehreren Prüfstellen durchgeführt,
ernennt der Sponsor einen Prüfer zum Leiter der klinischen Prüfung
[LKP]
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 25Ethik-Kommission
• Definition
– Unabhängiges Gremium bestehend aus Experten und Laien, das
sicherstellt, dass die Rechte, die Sicherheit und das Wohlergehen
der Prüfungsteilnehmer geschützt werden
• Nur ein Ethik-Votum pro Mitgliedsstaat (Richtlinie 2001/20/EG)
• In DE: Zuständig sind die nach Landesrecht gebildeten Ethik-
Kommissionen bei den
– Ärztekammern oder
– Medizinische Fakultäten
– z.Zt. 53 in DE
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 26Regulatorische Voraussetzungen
Betei- Betei- Betei- Betei-
ligte ligte ligte ligte
EK EK EK EK
Zustimmende
Bewertung der Genehmigung
zuständigen durch zuständige
Ethik- Bundesoberbehör
Kommission de
(BfArM oder PEI)
Beginn der
klinischen Prüfung
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 27Ethik-Kommissionen
Zuständige Ethik-Kommission Beteiligte Ethik-Kommissionen
• Verantwortlich für die • Verantwortlich für die
zustimmende Bewertung einer Geeignetheit von Prüfern und
klinischen Prüfung Prüfstellen
• Benötigen die Stellungnahme der • Arbeiten der zuständigen Ethik-
beteiligten EKs Kommission zu
• Verfahrensführend (Benehmensverfahren)
• Kommunikation mit dem Sponsor • Berufsrechtliche
sollte nur über die zuständige Beratungspflicht für Ärzte damit
Ethik-Kommission erfolgen abgegolten
• Zuständigkeit ergibt sich durch
den Sitz des Leiters der klinischen
Prüfung
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 28§ 40 AMG (1)
Allgemeine Voraussetzungen der klinischen Prüfung
• Zustimmende Bewertung der zuständigen EK und Genehmigung der
Bundesoberbehörde (BOB)
• Sponsor/Vertreter in der EU/EWR
• Positive Nutzen-/Risikoabwägung
• Anforderung an die betroffene Person (nicht in einer Anstalt)
• Anforderung an Aufklärung und Einwilligung
• Eignung der Prüfeinrichtung, Qualifikation der Prüfer
• Vorherige Durchführung einer pharmakologisch-toxikologischen Prüfung
• Information des Prüfers über die Ergebnisse durch einen
verantwortlichen Wissenschaftler
• Für medizinische Versorgung muss ein Arzt (ZA) verantwortlich sein
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 29§ 40 AMG (2)
Allgemeine Voraussetzungen der klinischen Prüfung
• Aufklärung durch Arzt (ZA)
– schriftliche Unterlagen, Rücktrittsrecht, Datenschutz …
• Versicherung
– haftungsunabhängig, Sitz des Versicherers, risikoadaptierter
Gesamtumfang, Einzelleistung bei Tod oder dauerhafter
Erwerbsunfähigkeit: 500.000 €, nicht zwingend erforderlich bei Phase IV-
Studien
• Prüfung an Minderjährigen
– Aufklärung, Einbeziehung des Willens des Betroffenen, gesetzlicher
Vertreter, „möglichst wenig Belastungen und Risiken“
• Einrichtung einer Kontaktstelle für betroffene Personen und gesetzliche
Vertreter bei der BOB
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 30§ 41 AMG
Besondere Voraussetzungen der klinischen Prüfung
• Absatz 1: Volljähriger Patient
– Individualnutzen oder Gruppennutzen
– Verzögerte Einwilligung in Notfallsituationen
• Absatz 2: Minderjähriger Patient
– Keine gesunden minderjährigen Probanden
– Individualnutzen oder Gruppennutzen
– „Minimal Risk – Minimal Burden“
• Absatz 3: Volljähriger aber nichteinwilligungsfähiger Patient
– NUR Individualnutzen - KEIN Gruppennutzen!
– Forschung muss sich auf einen lebensbedrohlichen oder sehr geschwächten
klinischen Zustand beziehen, in dem sich die betroffene Person befindet
– Geringe Belastung, Belastungsgrad muss im Prüfplan definiert sein und vom Prüfer
ständig überwacht werden
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 31§ 42 AMG
Verfahren bei der EK, Genehmigungsverfahren bei
der BOB
• Art und Umfang der vom Sponsor vorzulegenden Unterlagen
• Fristen für
– Bestätigung, Mängelbehebung, Bearbeitung
• Implizite Genehmigung bei Fristablauf durch BOB außer bei xenogenen
Zelltherapeutika
• Gegenseitige Unterrichtung von EK und BOB
• Regelung zu Aufgaben und Verantwortungsbereichen von Sponsor und
Prüfer durch Rechtsverordnung (GCP-Verordnung)
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 32Antragstellung
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 33Was muss eingereicht werden?
• Einreichung kann parallel bei BOB und EK erfolgen
• Unterlagen sind teilweise identisch für BOB / EK
• Wesentliche Unterlagen
– Studienprotokoll (Prüfplan)
– Anschriften aller Prüfstellen
– Investigator’s Brochure, IB (Prüferinformation)
– Dossier zum Prüfpräparat (IMPD)
• IMP = Prüfpräparat, IMPD=IMP-Dossier (nur BOB)
– Texte der Patienteninformation/ Patienteneinwilligungserklärung
(nur EK)
– Versicherungsunterlagen (nur EK)
– Wo beschrieben: § 7 GCP-V
• http://www.gesetze-im-internet.de/gcp-v/__7.html
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 34Inhalt eines IMP-Dossier (IMPD)
• Unterlagen über Qualität und Herstellung
• Vorgesehene Kennzeichnung (Labelling)
• Herstellungserlaubnis(se)
• Einfuhrerlaubnis(se)
• Unterlagen über die pharmakologisch-toxikologischen Prüfungen
– kann auch in der IB stehen
• Unterlagen über Ergebnisse von bisher durchgeführten klinischen
Prüfungen sowie weitere bekannt gewordene klinische Erkenntnisse
– kann auch in der IB stehen
• Zusammenfassende Nutzen-Risiko-Bewertung
– kann auch in der IB stehen
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 35Validierung Ablauf des Genehmigungsverfahrens Klinischer Prüfungen
Überprüfung auf Formmängel
Formaler Bescheid
Phase 1
ja nein
Inhaltliche Bewertung
einwandfrei?
Genehmigung Mit Gründen
versehene Einwände
Bescheid 1
Phase 2
nein
ja
Einwände ausgeräumt?
Bescheid 2
Versagung Genehmigung
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 36Kurze Fristen / Implizite Genehmigung
• 30 Tage Bewertungszeit für einen vollständigen Antrag
– Validierungsfrist von 10 Tagen enthalten
• Falls BfArM innerhalb der 30 Tage nach Einreichung keine
Entscheidung mitteilt gilt der Antrag als genehmigt
– Implizite Genehmigung!
BfArM
Sponsor
14/30 Tage 15d
90 Tage
Genehmigung Genehm.o.
oder Mängelbescheid Versagung
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 37Genehmigungsverfahren Klinischer Prüfungen im
BfArM
Pharm.
Präklinik Qualität Klinik
Verfahrensführung & Validierung
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 38Genehmigungs- Verfahrensführung
verfahren
Formale Prüfung Sachgebiet Sachgebiet
Sachgebiet
Klinische Pharmazeut.
Pharmakologie/Klinik Präklinik
Qualität
Formale Vollständigkeit
oder
formale Mängel
Inh. Prüfung 1 Sachgebiet
Sachgebiet Sachgebiet
Klinische
Präklinik Pharmazeut. Qualität
Pharmakologie/Klinik
Genehmigung oder
begründete Einwände
Inh. Prüfung 2 Ggf. Klinik Ggf. Präklinik Ggf. Pharm.Qualität
Finaler Bescheid
EudraCT-Eintrag PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 39Versagensgründe der BOB
§ 42 Abs. 2 AMG
• Unterlagen sind - auch nach Ablauf einer dem Sponsor gesetzten angemessenen
Frist zur Ergänzung – unvollständig
• Vorgelegte Unterlagen entsprechen nicht dem Stand der wissenschaftlichen
Erkenntnisse
• Prüfung insbesondere ungeeignet den Nachweis der Unbedenklichkeit oder
Wirksamkeit eines AMs, einschließlich Geschlechtsunterschieden, zu erbringen
• Nichterfüllung der Bedingungen nach §40 Abs. 1 Satz 3 Nr. 1, 2 und 6 (bei
xenogenen Zelltherapeutika auch Nr. 8)
• Der BOB liegen Erkenntnisse vor, dass die Prüfeinrichtung für die Durchführung
nicht geeignet ist oder dass von dieser die in Anforderungen an die klinische
Prüfung nicht eingehalten werden können
• Die § 40 Abs. 4 oder § 41 AMG sind nicht erfüllt
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 40Versagungsgründe: Nichterfüllung der
Bedingungen nach § 40 Abs. 1 Satz 3
BfArM/PEI EK
1. Fehlen eines Sponsors/Sponsorvertreters mit 2a. Falls GVO: GVO hat unvertretbaren
Sitz in der EU
schädlichen Auswirkungen auf Dritte
2. Die vorhersehbaren Risiken und Nachteile sind
und Umwelt
gegenüber dem Nutzen für die betroffenen
Personen und der voraussichtlichen Bedeutung 3. betroffene Person ist nicht geeignet
für die Heilkunde nicht ärztlich vertretbar sein (einwilligungsfähig, aufgeklärt,
2a. Falls GVO: GVO hat unvertretbaren schädlichen eingewilligt, Datenschutzaufklärung und
Auswirkungen auf Dritte und Umwelt -einwilligung liegt vor)
6. Keine vorherige Durchführung einer dem 4. betroffene Person ist auf gerichtliche
jeweiligen Stand der wiss. Erkenntnisse oder behördliche Anordnung in einer
entsprechende pharm./tox. Prüfung des AMs
Anstalt untergebracht
8. Versicherung deckt bei xenogenen Arzneimittel
* nicht auch die Risiken Dritter ab 5. Durchführung in ungeeigneter
Einrichtung oder nicht von angemessen
qualifizierten Prüfern durchgeführt.
7. Prüfer sind nicht über Pharm/Tox.
aufgeklärt worden
*Arzneimittel, die lebende tierische Gewebe oder Zellen sind oder enthalten 9. Medizinische Versorgung wird nicht
durch einen Arzt (oder ZA) verantwortet
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 41Praktische Antragstellung?
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 42Noch Fragen?
Ansprechpartnerin für organisatorische
oder administrative Belange:
Anna Zokol
Tel.: (0228) 207 – 3129
Anna.zokol@bfarm.de
Die Folien finden Sie unter: http://www.bfarm.de/DE/Service/Veranstaltungen/functions/_node.html
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 43Backup Slides
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 441000
2000
3000
4000
5000
6000
7000
8000
0
Germany
UK
Italy
France
Spain
Belgium
Netherland
Sweden
Czech Rep.
Austria
Hungary
Denmark
Poland
Finland
Norway
Slovakia
Portugal
Ireland
Greece
Lithuania
Bulgaria
Latvia
Estonia
Romania
Slovenia
Iceland
Malta
EudraCT: Clinical Trial Applications
Cyprus
1 May 2004 until 24 August 2010
Luxembourg
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 45Ethik-Kommission vs. BOB
Ethik-Kommission Bundesoberbehörde
• Beurteilung der Prüfung mit • Beurteilung der Prüfung mit
Schwerpunkten Schwerpunkten
Studiendurchführung und Studiendurchführung und
Belange des Prüfpräparat
Patienten/Probanden – Pharmazeutische
– Aufklärung Eigenschaften
– Datenschutz
– Finanzierung
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 46Genehmigung: Implizit / Explizit
• Erhebt die BOB nach Vorliegen eines ordnungsgemäßen Antrages keine gegründeten
Einwände innerhalb der vorgesehenen Frist, gilt die Klinische Prüfung als genehmigt
(implizite Genehmigung!)
– Aber: Sobald im Genehmigungsverfahren begründete Einwände erhoben wurden,
muss Sponsor auf explizite Genehmigung warten! (§9 GCP-V)
• Explizite Genehmigungen immer erforderlich bei AM,
– die unter die Nummer 1 des Anhangs der Verordnung (EG) Nr. 726/2004 fallen,
– die somatische Zelltherapeutika, xenogene Zelltherapeutika, Gentransfer-Arzneimittel
sind,
– die genetisch veränderte Organismen enthalten oder
– deren Wirkstoff ein biologisches Produkt menschlichen oder tierischen Ursprungs ist
oder biologische Bestandteile menschlichen oder tierischen Ursprungs enthält oder
zu seiner Herstellung derartige Bestandteile erfordert
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 47Nummer 1 des Anhangs der Verordnung (EG) Nr.
726/2004
• Arzneimittel, die mit Hilfe eines der folgenden biotechnologischen
Verfahren hergestellt werden:
– Technologie der rekombinierten DNS;
– kontrollierte Expression in Prokaryonten und Eukaryonten,
einschließlich transformierter Säugetierzellen, von
– Genen, die für biologisch aktive Proteine kodieren;
– Verfahren auf der Basis von Hybridomen und monoklonalen
Antikörpern
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 48Inhaltliche Prüfung
nein ja
Liegen Einwände
vor?
Implizites Verfahren
Zwischenbescheid:
Mit Gründen versehene
Einwände
Geänderte
Keine geänderten Unterlagen
Unterlagen
Explizites Verfahren
ja Einwände nein
ausgeräumt?
Finaler Bescheid: Finaler Bescheid:
Genehmigung des Antrags Versagung des Antrags
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 49Antrag auf nachträgliche
Änderung gemäß § 10 GCP-V
ja Antrag ordnungsgemäß? nein
(vollständig)
Finaler Bescheid:
Implizites Verfahren Ablehnung
nein ja
Begründete Einwände?
Finaler Bescheid: Finaler Bescheid: Begründete Einwände
Genehmigung Aufforderung zur Abänderung nach Maßgabe der BOB
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 50Beteiligte Behörden
Bundesoberbehörden (BOB) Behörden (Landesbehörden)
• BfArM und PEI • Multiple (ZLG)
• Direkt dem BMG unterstellt • Den Ländern unterstellt
• Genehmigungsbehörden des • Überwachungsbehörden des
AMG AMG
• Klinischen Prüfung • Klinische Prüfungen
– Genehmigung – Anzeige (Anfang / Ende)
– Sicherheitsüberwachung – Inspektionen
– Ergebnismitteilung
– Kommunikation mit der EU
– Inspektionen
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 51Vielen Dank für Ihre
Aufmerksamkeit!
Kontakt
Bundesinstitut für Arzneimittel und Medizinprodukte
Wissenschaftlicher Service
Kurt-Georg-Kiesinger-Allee 3
53175 Bonn
Ansprechpartner
PD Dr. Thomas Sudhop
Thomas.Sudhop@bfarm.de
www.bfarm.de
Tel. +49 (0)228 99 307-4360
Fax +49 (0)228 99 307-3424
PD Dr. Thomas Sudhop | Ringvorlesung- Einführung | 18.10.2016 | Seite 52Sie können auch lesen

















































