Monomeres C-reaktives Protein erniedrigt die Aufnahme von acetyliertem LDL in humane Endothelzellen
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Aus der Medizinischen Klinik und Poliklinik I
der Universität Würzburg
Direktor: Professor Dr. med. Georg Ertl
Monomeres C-reaktives Protein erniedrigt die Aufnahme
von acetyliertem LDL
in humane Endothelzellen
Inaugural-Dissertation
zur Erlangung der Doktorwürde der
Medizinischen Fakultät
der
Julius-Maximilians-Universität Würzburg
vorgelegt von
Matthias Christian Reichert
aus Nufringen
Würzburg, Januar 2010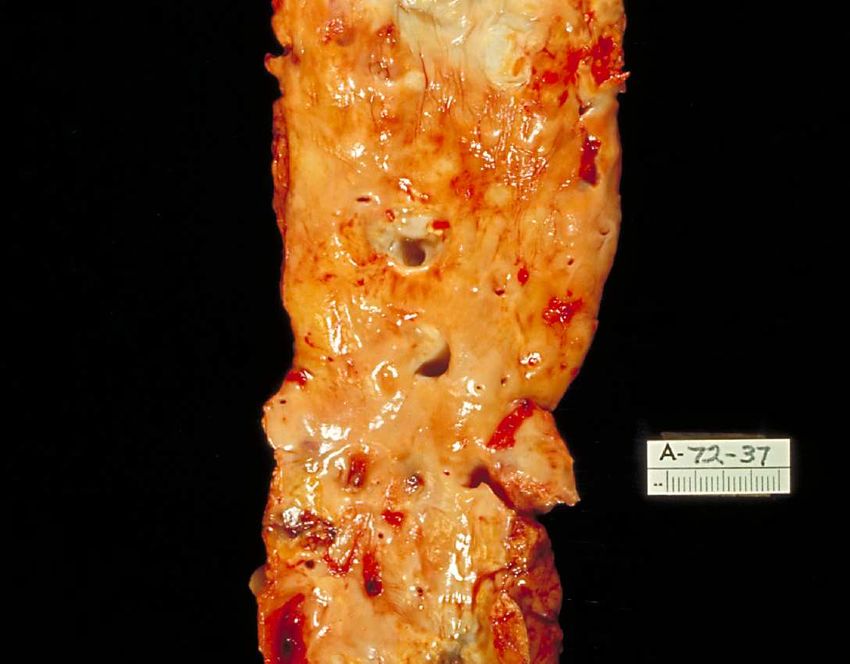
Referentin: Priv.-Doz. Dr. Susanne Schwedler Koreferentin: Prof. Dr. Michaela Kuhn Dekan: Prof. Dr. Matthias Frosch Tag der mündlichen Prüfung: 26.05.2010 Der Promovend ist Arzt
Meinen Eltern
Inhaltsverzeichnis
1. Einleitung 1
1.1 Abkürzungen 1
1.2 Arteriosklerose: Epidemiologie und Folgeerkrankungen 3
1.3 Pathogenese der Arteriosklerose 4
1.3.1 Die response-to-injury-Hypothese 5
1.3.2 Die Lipoprotein-induzierte-Arteriosklerose-Hypothese 9
1.4 Low density lipoprotein (LDL) und modifiziertes low density 9
-lipoprotein (mLDL) in der Arteriosklerose
1.4.1 Low density lipoprotein (LDL) 9
1.4.2 Zelluläre Aufnahme von LDL 10
1.4.3 Oxidiertes LDL (oxLDL) 10
1.4.4 Acetyliertes LDL (acLDL) als Modell für oxidiertes LDL 11
1.5 C-reaktives Protein (CRP) 12
1.5.1 CRP: Struktur 13
1.5.2 CRP: Konfigurationen 14
1.5.3 CRP: Regulation 15
1.5.4 CRP: Liganden 15
1.5.5 CRP: Interaktion mit LDL 17
1.6 CRP und Arteriosklerose 17
1.6.1 Die CRP-Konfigurationen in der Arteriosklerose 19
1.7 CRP-Rezeptoren 20
1.7.1 CD16 (FcγRIII) 21
1.7.2 CD32 (FcγRII) 21
1.7.3 CD64 (FcγRI) 22
1.7.4 Zusammenfassung 22
1.8 Fragestellung 23
I
2. Material und Methoden 24
2.1 Chemikalien und Reagenzien 24
2.1.1 Antikörper 25
2.1.2 C-reaktives Protein (CRP) 25
2.1.2.1 Natives CRP (nCRP) 25
2.1.2.2 Modifiziertes CRP (mCRP) 26
2.1.3 Lipoproteine 26
2.1.4 Endothelzellmodell: HUVEC 28
2.2 Geräte und Materialien 29
2.3 Zellkultur 30
2.3.1 Inkubation 31
2.3.2 Durchflusszytometrische Analyse 31
2.4. Immunhistochemie 32
2.5 Molekulargenetische Untersuchungen 33
2.5.1 RNA-Isolierung 33
2.5.2 RT-PCR 33
2.6 Statistik 34
3. Ergebnisse 35
3.1 Effekte von modifiziertem und nativem CRP mit/ohne oxLDL auf die acLDL 35
Aufnahme
3.2 Lokalisation von mCRP, nCRP und acLDL in HUVEC 40
3.3 Effekte von modifiziertem und nativem CRP mit/ohne ox LDL auf CD16 43
und CD32
4. Diskussion 44
5. Zusammenfassung 51
6. Literaturverzeichnis 52
7. Abbildungsverzeichnis 65
7.1 Abbildungen 65
7.2 Tabellen 66
8. Publikationen I
9. Danksagung II
II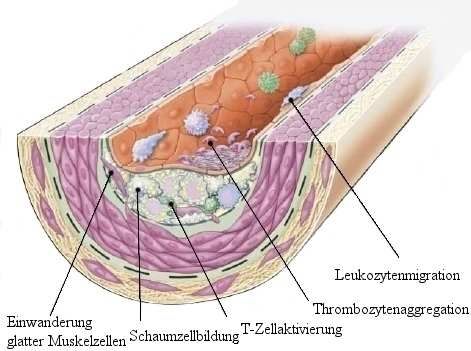
Einleitung
1. Einleitung
1.1 Abkürzungen
acLDL acetyliertes LDL
ApoE Apolipoprotein E
AT-1 Angiotensin 1
CRP C-reaktives Protein
CK Creatininkinase
CK-MB Creatininkinases-musclebrain
DiL 1,1’-Dioctadecyl-1-3,3,3’,3’-tetramethyl-indocarbocyanin-perchlorat
E-LDL enzymatisch modifiziertes LDL
dil-ac-LDL fluoreszenzmarkiertes (Dil) acetyliertes low density lipoprotein
eNOS endotheliale NO-Synthase
FCS fetal calf serum
HDL High-density Lipoprotein
HAEC Human aortic endothelial cells
HCAEC Human coronary artery endothelial cells
HUVEC Human umbilical vein endothelial cells
ICAM intercellular adhesion molecule
IgG Immunglobulin G
IκB inhibitorisches κB
IL-1,6,8,10 Interleukine 1,6,8,10
iNOS induzierbare NO-Synthase
LDL Low-density Lipoprotein
LPS Lipopolysaccarid
MAPK mitogen activated protein kinase
MCP-1 monocyte chemoattractant protein 1
mCRP Modifiziertes/monomeres CRP
MMP-1 matrix metalloproteinase-1
nCRP natives CRP
NFκB nuclear factor қB
nLDL natives LDL
NO Stickstoffmonoxid
1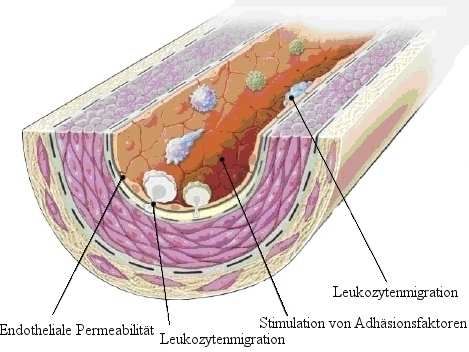
Einleitung
oxLDL oxidiertes LDL
PAI plasminogen activator inhibitor
PDGF platelet derived growth factor
TNF-α Tumornekrosefaktor α
VCAM vascular cell adhesion molecule
VEGF vascular endothelial growth factor
VLDL very low-density Lipoprotein
2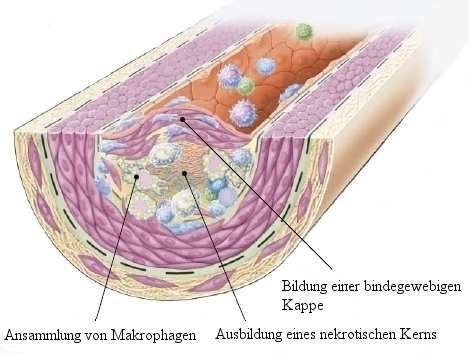
Einleitung
1.2 Arteriosklerose: Epidemiologie und Folgeerkrankungen
Arteriosklerose (griechisch „Verhärtung der Arterien“) bezeichnet die krankhafte
Veränderung der Arterien mit Verhärtung, Verdickung, Elastizitätsverlust und Einengung des
Gefäßlumens1. Der Befall der großen, mittleren und kleineren Gefäße wird als
Makroangiopathie, Befall sehr kleiner Gefäße als Mikroangiopathie bezeichnet.
Weltweit ist die Arteriosklerose mit ihren Folgeerkrankungen die häufigste Todesursache
und eine häufige Ursache für Morbidität (Abb. 1), wobei die westlichen Industrienationen
besonders betroffen sind. Aber auch die Entwicklungsländer haben inzwischen
aufgeschlossen.
Abb. 1 Todesfälle weltweit 2005, sortiert nach Ursachen für alle Altersgruppen
Adaptiert nach: WHO Chronic diseases and health promotion: preventing chronic disease:
A vital investment. 2005
http://www.who.int/chp/chronic_disease_report/en
Arteriosklerose ist ein multifaktorieller Prozess, der im frühesten Alter beginnt und bereits
in fetalen Aorten2 und in Koronargefäßen von Jugendlichen und jungen Erwachsenen3
nachgewiesen werden konnte, sich aber klinisch in der Regel ohne Vorhandensein von
Gendefekten erst in späteren Lebensabschnitten manifestiert. Die wichtigsten klassischen
Risikofaktoren wurden in einer großangelegten Studie die 1948 in Framingham in den USA
begonnen wurde und aktuell noch einschließlich der Enkelgeneration weiter läuft erkannt
(Framinghamkohorte). Dazu zählen Hyperlipidämie, Hypercholesterinämie, Hyperurikämie,
LDL-Überschuss, HDL-Mangel, arterielle Hypertonie, Adipositas, Rauchen, Diabetes
mellitus, ungesunde Ernährung, Bewegungsmangel, psychosoziale Faktoren und die
obstruktive Schlafapnoe. Inzwischen sind insgesamt über 300 Risikofaktoren für die
3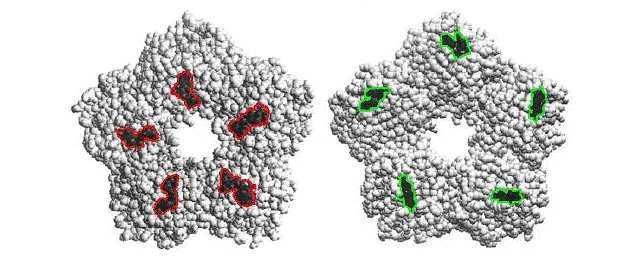
Einleitung
Entwicklung von Arteriosklerose und ihrer Folgeerkrankungen bekannt geworden4. In den
letzten 10 Jahren wurden zahlreiche klinische Studien veröffentlicht, die CRP als weiteren
wichtigen Risikofaktor in der Arteriosklerose identifizieren. So konnte eine Korrelation
zwischen dem CRP-Blutspiegel und der Rate an kardiovaskulären Ereignissen festgestellt
werden5,6,7. Tierexperimentelle Arbeiten wiesen CRP darüber hinaus auch eine Rolle als
Mediator bei der Entstehung der Arteriosklerose zu8. Arteriosklerose ist ein Prozess, der in
Stadien beschrieben werden kann (siehe Kapitel 1.3). Im schlimmsten Fall führt er zur Ruptur
eines sogenannten Plaques, was dann zur Thrombose und zur Beeinträchtigung der arteriellen
Gefäßversorgung und nachfolgender Ischämie führt. Viele Erkrankungen beruhen auf
arteriosklerotischen Veränderungen: Myokardinfarkt, ischämischer Hirninfarkt, Aneurysmen,
Lungenembolien, Niereninsuffizienz, Herzklappenerkrankungen und Durchblutungsstörungen
in Extremitäten, Darm, Niere und Auge.
1.3 Pathogenese der Arteriosklerose
Arteriosklerotische Läsionen sind asymmetrische Verdickungen der arteriellen Intima.
Die Gefäßwand enthält zahlreiche Zellen wie Lymphozyten, Makrophagen, Schaumzellen,
sogenannte dendritische Zellen, Mastzellen und Bindegewebe, Blutgefäße, glatte
Muskelzellen sowie Zellschrott.
Eine kausale Beziehung zwischen Cholesterinspiegel im Serum und Arteriosklerose ist
inzwischen allgemein anerkannt, erhöhte Cholesterinspiegel sind jedoch nur Teil der
Pathogenese der Arteriosklerose und können alleine deren Enstehung nicht erklären.
Arteriosklerose wird in den letzten Jahren zunehmend als immunsystemvermittelter,
entzündlicher Prozess der Gefäße angesehen9,10. Entzündung ist ein komplexer Prozess, in
dem Entzündungs- und Immunzellen, vor allem Leukozyten, nach einem Stimulus die Gefäße
verlassen und ins Gewebe einwandern. In der Arteriosklerose ist der auslösende Stimulus für
diesen Prozess noch nicht bekannt, unter anderem werden oxidiertes LDL (oxLDL) und
minimal oxidiertes LDL diskutiert10.
4
Einleitung
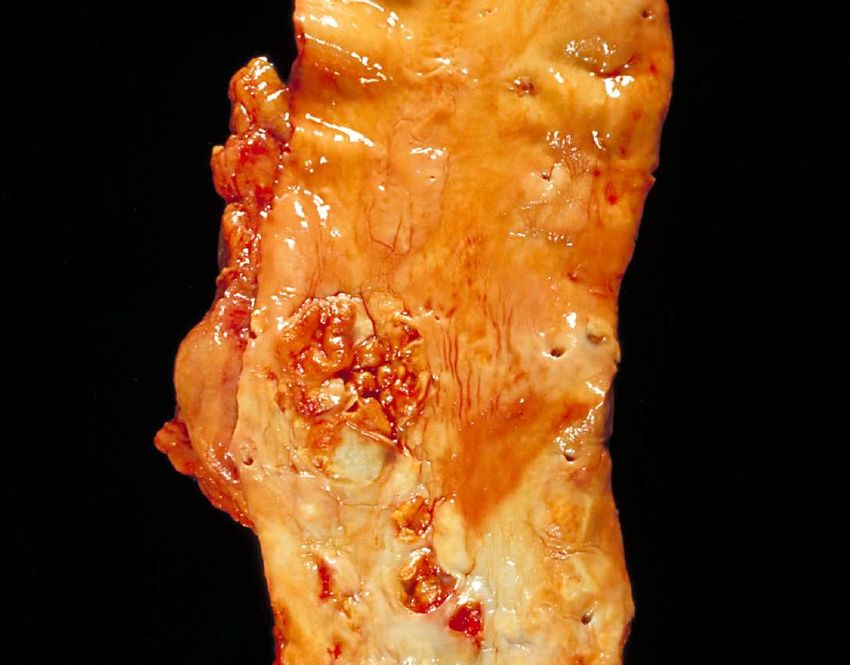
Abb. 2 Menschliche Aorta längs aufgeschnitten mit fortgeschrittenen arteriosklerotischen Veränderungen wie
Plaques mit zahlreichen Ulzerationen
Public Health Image Library, Centers for Disease Control and Prevention, 1600 Clifton Road
Atlanta, GA 30333, USA /Dr. Edwin P. Ewing, Jr.
1.3.1 Die response-to-injury-Hypothese
Zahlreiche pathophysiologische Beobachtungen in Mensch und Tier führten zur response-to-
injury Hypothese, formuliert von Ross 1976. Sie besagt, dass der erste Schritt in der
Pathogenese die endotheliale Dysfunktion ist, und die körpereigene Reaktion auf diesen
Schaden dann entscheidend für die weitere Entwicklung der Arteriosklerose ist9. Zu den
Auslösern für eine endotheliale Dysfunktion gehören Dyslipidämien (erhöhte LDL-
Blutspiegel und modifizierte LDL-Proteine), freie Radikale (beispielsweise induziert durch
Zigarettenrauchen), Hypertonie, genetische Veränderungen und infektiöse Mikroogranismen,
wie Herpesviren oder Chlamydia pneumoniae11,12. Mit endothelialer Dysfunktion wird die
Aktivierung des Endothels aus einem ruhenden Zustand bezeichnet. Sie führt zu verminderter
endothelvermittelter Vasodilatation. Auf molekularer Ebene ensteht dies durch ein
Ungleichgewicht mit Abnahme von vasodilatatierenden und Zunahme von
vasokonstringierenden Faktoren und Botenstoffen. Wichtige vasodilatatierenden Substanzen
im Endothel sind NO und Prostazyklin. Vasokonstriktorisch wirkenden Substanzen sind
Endothelin 1, Angiotensin II und Plasminogen activator inhibitor-1 (PAI-1)8 (Abb. 3).
Zusätzlich ist die endotheliale Dysfunktioin durch ein Überwiegen proinflammatorischer und
prothrombotischer Eigenschaften des Endothels charakterisiert13.
5Einleitung
Abb. 3 Endotheliale Dysfunktion
Die frühsten Veränderungen finden sich im Endothel: Erhöhte endotheliale Permeabilität für
Lipoproteine und andere Blutplasmabestandteile, vermittelt durch Botenstoffe wie NO,
Prostazyklin, PAI-1, Angiotensin II und Endothelin 1. Leukozytenadhäsionsmoleküle wie L-
Selectin, Integrine und endotheliale Adhäsionsmoleküle wie E-Selectin und P-Selectin werden
heraufreguliert. Leukozyten wandern vermittelt durch oxLDL, MCP-1, Interleukin-8, PDGF,
macrophage colonystimulating factor und andere ein
Aus Ross et al9
Eine wichtige Rolle in der Entstehung der Arteriosklerose spielen die modifizierten
Lipoproteine. In der endothelialen Dysfunktion ist die Permeabilität des Endothels für
Lipoproteine und andere Blutplasmabestandteile erhöht. Eingewanderte Lipoproteine können
bei längerer Verweildauer im subintimalen Spalt oxidativen Modifikationsprozessen der
Gefäßwand ausgesetzt werden. Hierbei kommt es an Prädilektionsstellen der Arterien
(Gefäßabgänge, Richtungsänderungen) zu einer verlängerten Retentionszeit der Lipoproteine.
Ausgelöst wird dies durch erhöhte endotheliale Permeabilität und durch Bindung an
Bestandteile der extrazellulären Matrix. Es entstehen Lipoproteinaggregate, die die Intima
nicht mehr verlassen können und von Enzymen in der Arterienwand oxidiert werden. OxLDL
und die an es gebundenen Proteine verfügen über neue Epitope, gegen die eine
Entzündungsreaktion stattfinden kann. Nach Exposition mit oxLDL erhöhen Endothelzellen
in der Gefäßwand ihre Expression von Adhäsionsmolekülen und Chemokinen wie VCAM-1
oder MCP-1, was Monozyten und Leukozyten anlockt. Dies führt zur Adhärenz der Zellen an
das Endothel und zur Migration in die Intima. In der Media erfolgt dann eine Differenzierung
der Monozyten zur Makrophagen aufgrund des lokal produzierten Makrophagen-colony-
stimulating factors (M-CSF). Die Makrophagen nehmen oxLDL über Scavenger-Rezeptoren
konzentrationsunabhängig auf. Diese sind nicht in der Lage die Fülle an Cholesterin zu
verstoffwechseln, sondern lagern es in Form von Cholesterinestertröpfchen intrazellulär ab.
6Einleitung
Mikroskopisch haben diese Zellen nach Entfettung des Präparats ein schaumiges Aussehen
und werden daher auch Schaumzellen genannt. Diese wiederum verursachen eine
Entzündungsreaktion, welche im Verlauf auf tiefere Bereiche der Arterienwand wie der
Media mit ihren Muskelzellen übergreifen kann. Dort können dann auch die glatten
Muskelzellen Cholesterinester aufnehmen und zu Schaumzellen werden (Abb. 4).
Abb. 4 Bildung von Fettstreifen (“fatty streaks”)
Fettstreifen bestehen zunächst aus Lipidbeladenen Monozyten und Makrophagen
(Schaumzellen) und T-Lymphozyten. Später kommen glatte Muskelzellen hinzu. An der
Entwicklung beteiligt ist die Migration von glatten Muskelzellen, stimuliert durch den
plättchenabhängigen Wachstumsfaktor (PDFG), Fibroblasten Wachstumsfaktor 2 und
transforming growth factor β. T-Zellen werden aktiviert , es kommt zur Schaumzellbildung und
Thrombozytenanlagerung und -aggregation
Aus Ross et al9
Nun beginnt ein Teufelskreis: Die neu entstandenen Schaumzellen schütten
proinflammatorische Zytokine aus, die dann zur Bildung von reaktiven Sauerstoffradikalen
führen und die endotheliale Dysfunktion verstärken9,14. Die Sauerstoffradikale führen zur
Verminderung der NO-Aktivität, stimulieren das Wachstum von glatten Muskelzellen,
initiieren die Expression von proinflammatorischen Genen15 und oxidieren weiter
Lipoproteine. Wenn es das Immunsystem nicht schafft die auslösenden Faktoren zu
eliminieren, führt dies zur Chronifizierung des entzündlichen Prozesses. Die Gefäßwand wird
umstrukturiert und verdickt. Noch kann dies aber durch Dilatation des Gefäßes ausgeglichen
werden. Dieser Prozess wird Remodelling genannt. Beteiligt an diesem Prozess sind vor allem
Makrophagen und T-Lymphozyten, die sich bei chronischen Entzündungen vermehren und
verstärkt aus dem Blut in die Gefäße einwandern16. Diese Zellen setzten hydrolytische
7Einleitung
Enzyme, Zytokine, Chemokine und Wachstumsfaktoren frei, die zur Verstärkung des
Schadens und zur fokalen Nekrose führen können. Monozyten und Makrophagen sammeln
sich an, glatte Muskelzellen wandern ein und proliferieren. Es wird Bindegewebe gebildet, die
Läsion wird weiter vergrößert und umstrukturiert. An kleinsten Verletzungen am Endothel
anhaftende Thrombozyten sezernieren ebenfalls Wachstumsfaktoren (unter anderem PDGF),
was glatte Muskelzellen aus der Media anlockt und deren Proliferation fördert17. Diese glatten
Muskelzellen haben kaum noch kontraktile Eigenschaften, sezernieren aber zusammen mit
den ebenfalls vorhandenen Fibroblasten extrazelluläre Matrix und Kollagen. So entsteht eine
bindegewebige Kappe, die einen Kern aus Lipiden und nekrotischem Gewebe bedeckt – eine
komplizierte Läsion (Abb. 5). Im Lipidkern befinden sich abgestorbene Schaumzellen, die
große Mengen an oxLDL aufgenommen haben. Eine solche fortgeschrittene Läsion enthält T-
Lymphozyten (~10%), Makrophagen und Schaumzellen (~40%), glatte Muskelzellen,
vaskuläre Zellen, einige wenige B-Lymphozyten, dendritische Zellen, Mastzellen und
sogenannte "Natürliche Killerzellen"18.
Abb. 5 Entwicklung einer komplizierten Läsion
Fettstreifen entwickeln sich zu komplizierten Läsionen und bilden eine bindegewebige Kappe
aus, die sich ins Lumen des Gefäßes vorwölbt. Die bindegewebige Kappe bedeckt eine
Ansammlung von Leukozyten, Lipiden und Zellschrott, der einen nekrotischen Kern bilden
kann. Die Läsion beginnt sich zu vergrößern und ins Gefäßlumen einzudringen
Aus Ross et al9
Ab einem gewissen Punkt ist die Einengung des Lumens der Arterie nicht mehr durch
Dilatation kompensierbar und die Läsion dringt ins Lumen des Gefäßes ein. Dies kann den
Blutfluss behindern. Eventuell ulzerieren und brechen die Plaques auf. Es besteht dann die
Möglichkeit, dass sich an den Plaques Blutgerinnsel bilden. Eine weitere Einengung des
Lumens und schließlich die Bildung eines Thrombus mit nachfolgender Ischämie des
Endstromgebiets der spezifischen Arterie kann dann folgen. Eine Lösung von Teilen des
8Einleitung
Thrombus als Embolus mit unter Umständen konsekutivem Gefäßverschluss an anderer Stelle
ist eine weitere potentielle Gefahr.
1.3.2 Die Lipoprotein-induzierte-Arteriosklerose-Hypothese
Im Laufe der Zeit zeigte sich, dass die endotheliale Dysfunktion, die in der „response-to-
injury-Hypothese“ für die Entstehung der Arteriosklerose verantwortlich gemacht wird, nur
einen Teil der komplexen Pathogenese der Arteriosklerose erklären kann. Eine neue
Hypothese, die besagt, dass die endotheliale Dysfunktion nur ein Teilschritt einer spezifischen
Form der chronischen Entzündung ist, wurde entwickelt. Die Interaktion von
Plasmalipoproteinen und zellulären Komponenten wie Monozyten/Makrophagen, T- und B-
Lymphozyten, Endothel- und glatten Muskelzellen sowie der extrazellulären Matrix der
Arterienwand und CRP wird hier in einem komplexen Zusammenspiel für die Entstehung der
Arteriosklerose verantwortlich gemacht. In den modifizierten Lipoproteinen wie oxLDL wird
hier die eigentliche Ursache für die Initiierung des arteriosklerotischen Geschehens gesehen
(Lipoprotein-induzierte-Arteriosklerose-Hypothese).
1.4 Low density lipoprotein (LDL) und modifiziertes low density lipoprotein (mLDL) in der
Arteriosklerose
1.4.1 Low density lipoprotein
Low Density Lipoproteine (LDL) gehören zur Klasse der Lipoproteine. Sie dienen als
Transportvehikel für lipophile Substanzen wie Cholesterin, Cholesterinester, Triglyceride,
Fettsäuren und Phospholipide sowie lipophile Vitamine, etwa Vitamin E und Vitamin A.
Humanes LDL hat eine Dichte von 1,019 bis 1,062 g/ml und eine Größe von 18 bis 25 nm.
Es besteht aus einem Apolipoprotein B100 (Apo B100) mit einer molaren Masse von 550 kDa
und ungefähr 1600 Molekülen Cholesterinestern, 600 Molekülen freies Cholesterin, 700
Molekülen Phospholipiden und 185 Molekülen Triglyceride19. Die Rolle von LDL und des in
ihm enthaltenen Cholesterins als Risikofaktor für die Arteriosklerose sind aus der
Framinghamkohorte schon seit 1977 bekannt20, die Bedeutung von oxidativ modifziertem
LDL in der Entstehung der Arteriosklerose hingegen erst einige Jahre später.
9Einleitung
1.4.2 Zelluläre Aufnahme von LDL
LDL kann in allen zellkernhaltigen Zellen über den LDL-Rezeptor aufgenommen werden.
Der LDL-Rezeptor ist ein transmembranöses Protein, das durch Endozytose die Aufnahme
von LDL vermittelt21. Der LDL-Rezeptor erkennt in der Phospholipidschicht von LDL
Apoprotein B100. Er erkennt auch das ApoE-Protein in Chylomikronen und VLDL. Entdeckt
wurde er 1985 von den späteren Nobelpreisträgern Brown und Goldstein22. Defekte führen
zur familiären Hypercholesterinämie. In den späten 80er Jahren begann klar zu werden, dass
LDL in die entstehenden arteriosklerotischen Plaques und Schaumzellen vermutlich nicht
über den von Brown und Goldstein entdeckten LDL-Rezeptor aufgenommen wird, sondern
über einen alternativen, noch unbekannten Rezeptor. Dies wurde aus der Beobachtung
geschlossen, dass Patienten und Versuchstiere auch ohne LDL-Rezeptor Cholesterin einlagern
können, und in Monozyten/Makrophagen selbst bei hohen LDL-Konzentrationen nur geringe
Mengen an Lipoproteinen aufgenommen werden können. Die Modifizierung von LDL durch
Acetylierung ermöglichte eine hohe LDL-Aufnahme in Makrophagen23. Es wurde
geschlussfolgert, dass es eine modifizierte LDL-Form gibt, die durch einen spezifischen
Rezeptor aufgenommen werden kann. Hinzufügen von Antioxidantien zum Kulturmedium
blockierte die Aufnahme von Cholesterin. Zugabe von oxidierenden Metallen verstärkte die
Aufnahme, sodass eine oxidativ modifizierte LDL-Form für einen großen Anteil der
übermäßigen LDL-Aufnahme verantwortlich gemacht wurde. Dieser Rezeptor wurde
identifiziert und Scavengerrezeptor A (SRA) benannt24. Später wurden noch weitere
Scavengerrezeptoren wie Scavengerrezeptor B, CD36, MACRO, Macrosialin (CD68)25 und
LOX-1 entdeckt. LOX-1 wird heute als wichtigster Rezeptor für oxLDL in Endothelzellen
angesehen26,27.
1.4.3 Oxidiertes LDL (oxLDL)
Die Bildung von reaktiven Sauerstoffspezies ist Teil eines unspezifischen Abwehrsystems des
Körpers gegen Mikroorganismen und Fremdkörper, wie z.B. Bakterien. Reaktive
Sauerstoffspezies, wovon die wichtigsten Superoxid O2-, Hydrogenperoxid H2O2, Peroxinitrit
ONOO- und Stickoxid NO sind, können jedoch auch Zellen des eigenen Körpers angreifen,
vor allem an Stellen wo
10Einleitung
Entzündungsaktivität vorhanden ist. Reaktive Sauerstoffspezies werden durch die NAD(P)H-
Oxidase, Lipoxygenase, Mitochondrien oder die NO-Synthase produziert. Es gibt zahlreiche
Auslöser für die Bildung von reaktiven Sauerstoffspezies, unter anderem Katecholamine,
physischer Stress und Entzündungsmediatoren wie CRP und Tumornekrosefaktor α (TNF-α).
Vor allem im subintimalen Gefäßspalt kommt es zur Oxidation von LDL-Aggregaten durch
Enzyme in der Arterienwand wie Myeloperoxidase oder Lipoxygenase. Oxidierte
Lipoproteine können auch in apoptotischen Zellen und Entzündungen entstehen. Oxidierung
von LDL selbst erhöht die Bildung von 02-. Natives, nichtoxidiertes LDL hingegen hat kaum
Einfluss auf die Bildung von reaktiven Sauerstoffspezies27. OxLDL induzierte O2-Bildung
führt zu endothelialer Dysfunktion und entweder Zellwachstum oder Apoptose und Zelltod28
und trägt so zum Remodelling der Gefäßwand bei. OxLDL kann die Expression von
Adhäsionsmolekülen und Chemokinen auf dem Endothel induzieren (unter anderem
Endothelin-1, MCP-1, VCAM-1, Aktivierung des NFκB-Signalweges) und so unter anderem
die Migration von Monozyten und Leukozyten in die Intima verstärken. Zusätzlich aktiviert
oxLDL den vasokonstriktorischen kontraktilen Apparat der Gefäßwand29. So vermittelt
oxLDL endotheliale Dysfunktion und führt zum Remodelling der Gefäßwand. Der Schaden
von oxLDL im Fortschreiten der Arteriosklerose geht über den der erhöhten Aufnahme
hinaus.
1.4.4 Acetyliertes LDL (acLDL) als Modell für oxidiertes LDL
Das am häufigsten in der Literatur verwendet Modell zur Untersuchung von oxLDL in der
Zellkultur ist acetyliertes LDL (acLDL). Da ox- und acLDL eine überlappende
Rezeptorbindungskapazität haben, ist es möglich durch die Behandlung von LDL mit
Essigsäureanhydrid acLDL zu generieren, das durch SRA erkannt und aufgenommen werden
kann. Dafür ist es nötig, dass mindestens 60% der Lysinaminogruppen des LDLs maskiert
sind19 Die Acetylierung mit Essigsäureanhydrid führt durch eine Veränderung in der
Proteinladung und der räumlichen Konfigurationen zur Maskierung der Lysin ε-
Aminogruppen. Acetyliertes LDL wurde von uns verwendet, um die Aufnahme von
modifiziertem LDL in Endothelzellen ohne oxidativen Stress untersuchen zu können.
11Einleitung
1.5 C-reaktives Protein (CRP)
C-reaktives Protein (CRP) wurde schon 1930 von Tillet und Francis im Blut von Patienten
mit einer Streptokokkus pneumoniae-Infektion entdeckt. Es kann das C-Polysaccarid der
Zellwand der Pneumokokken präzipitieren und wurde deshalb als C-reaktive Substanz
bezeichnet30. Später wurde die Substanz in C-reaktives Protein umbenannt31. CRP ist ein in
der Leber synthetisiertes Akut-Phase-Protein mit einem Molekulargewicht von 11800 kDa.
Die Konzentration im Serum kann zytokinabhängig innerhalb von 24 bis 72 Stunden bis auf
das 1000fache des Normwertes (0,1-1 mg/dl) ansteigen und spiegelt bei Infektionen,
Gewebsverletzungen und Entzündungen die Schwere und den Verlauf der Erkrankung
beziehungsweise der Verletzung wieder. Das im Serum gemessene, pentamere CRP wird auch
als natives CRP (nCRP) bezeichnet. Es hat eine Halbwertszeit von ungefähr 19 Stunden.
Klinisch wird es vor allem zur Überwachung und Entdeckung von akuten Infektionen sowie
zur Kontrolle von chronisch entzündlichen Erkrankungen, wie Vaskulitiden und rheumatoider
Arthritis verwendet. Die Fähigkeit von CRP pathogene Substanzen zu erkennen, zu markieren
und zu zerstören macht es zu einem wichtigen Bestandteil des angeborenen Immunsystems,
insbesondere bei der Abwehr von bakteriellen Infekten und der Zerstörung beschädigter
Zellen. CRP ist ein phylogenetisch hochkonserviertes Protein mit homologen Proteinen in
Wirbeltieren und vielen Wirbellosen, wie beispielsweise dem Pfeilschwanzkrebs Limulus
polyphemus, der seit 70 Millionen Jahren existiert. Dies und die Tatsache, dass man in der
Struktur von CRP nur wenige Polymorphismen finden konnte, lässt vermuten, dass es eine
lebenswichtige Rolle spielt31. CRP-Genpolymorphismen haben einen Einfluss auf die Höhe
des Blutspiegels. So konnten Wang et al in einer Studienkohorte zeigen33, dass Single-
nucleotid-polymorphismen (SNPs) einen Einfluss auf die Höhe des Blutserum-CRP-Spiegels
haben. Dort zeigte sich eine hochsignifikante Assoziation von SNP 757 und SNP 286 sowie
grenzwertige Assoziation von SNP 7180 mit erhöhten CRP-Blutspiegeln. Im CRP-Spiegel
zeigen sich geschlechts- und rassenspezifische Unterschiede. Schwarze haben höhere CRP-
Spiegel als Weiße, und Frauen haben höhere als Männer34. Der CRP-Spiegel ist auch erhöht
bei Rauchern, im Alter, bei malignen Erkrankungen, bei Infektionen und Entzündungen,
Niereninsuffizienz und Dialyse, durch Adipositas, metabolisches Syndrom, Typ 2 Diabetes,
Bluthochdruck und obstruktiver Schlafapnoe.
12Einleitung
1.5.1 CRP: Struktur
CRP gehört zu den sogenannten Pentraxinproteinen. Es ist ein sehr gut wasserlösliches
Serumprotein, das calciumabhängige Affinität für Phosphatmonoester aufweist, vor allem
Phosphatidylcholin. Die molekulare Struktur von CRP kann mit Hilfe der
Röntgenkristallographie in einer Auflösung von 3Å dargestellt werden35. Die Struktur des
CRP hat einen Außendurchmesser von 102Å mit einer zentralen Pore mit einem Durchmesser
von 30Å. Die Struktur ist ein kreisförmiger Diskus (Pentamer) aus fünf, nicht-kovalent
miteinander verbundenen identischen Untereinheiten (Monomeren) mit einem Durchmesser
von jeweils 36Å (Abb. 6). Die Monomere bestehen aus 206 Aminosäuren (23kDa) die in 14
β-Strängen antiparallel zueinander stehen. Diese sind wiederum in zwei β-Faltblättern
angeordnet. Alle Monomere sind im CRP-Molekül symmetrisch um die zentrale Pore
angeordnet, die beiden Seiten des Moleküls unterscheiden sich, sie werden unterteilt in die A-
und B-Seite. Die A-Seite verfügt über C1q- und Fcγ-Rezeptor-Bindungsstellen. Diese
befinden sich allesamt in den Spalten, die sich durch die Faltung des Monomers ergeben. Die
B-Seite beherbergt die Phosphatidylcholin–Bindungsstelle und zwei Bindungsstellen für
Calciumionen pro Monomer (Abb. 7).
Abb. 6 Pentamere Struktur des CRP. Links gezeigt A-Seite des nativen CRP (nCRP). Rechts ein
Monomer (mCRP)
Aus Shrive et al35
13Einleitung
A-Seite B-Seite
Abb. 7 Molekulares Modell von CRP, basierend auf der Brookhaven Protein Data Base
A-Seite: Fcγ-Rezeptor Bindungsstelle rot umrandet
B-Seite:Calciumabhängige Phosphatidylcholinbindungsstelle grün umrandet
Nach Marnell et al36
1.5.2 CRP: Konfigurationen
1988 wurden durch Samberg et al in Lymphozyten neue Epitope, die auf dem nativen CRP
nicht vorhanden waren, entdeckt, die nach Dissoziation der fünf Untereinheiten von nCRP
entstanden. Dieses „neo-CRP“ konnte nur noch teilweise durch anti-CRP-Antikörper erkannt
werden37 und erhielt später die Bezeichnung „modifiziertes C-reaktives Protein“ (mCRP).
Zerfällt das Pentamer nCRP in seine Monomere, kommt es zu einer Konformationsänderung,
zu einer Abnahme der Wasserlöslichkeit und zur Expression neuer Epitope. Deshalb wird es
auch monomeres CRP genannt. Die Löslichkeit von mCRP ist erniedrigt. Diese Umwandlung
von nCRP in mCRP erfolgt nicht proteolytisch und ist irreversibel. Modifiziertes CRP kann
durch Konversion aus nCRP durch Harnstoffchelatierung, Hitze, Säuren, Kontakt mit
Zellmembranen39 oder direkte Immobilisierung auf Polystyrenplatten entstehen39,40. Längere
Lagerung von gereinigtem CRP ohne Calcium oder Chelatoren führt zur spontanen
Konversion von nCRP zu mCRP41. Über die Faktoren der Konversion in vivo wird noch
spekuliert. Modifiziertes CRP ist ein wahrscheinlich im menschlichen Organismus
physiologisch vorkommendes Protein. So fanden sich Ablagerungen von mCRP in
Makrophagen/Monozyten42, Lunge43,44 und den Gefäßen zahlreicher Gewebe wie Herz44,
Ovar44, Hoden44 und Lunge43,44. Modifiziertes CRP konnte jedoch bisher nicht im Blutserum
nachgewiesen werden. Der immunhistochemische Nachweis wurde ermöglicht durch die
Herstellung zahlreicher monoklonaler Antikörper, mit deren Hilfe nCRP von mCRP
unterschieden werden kann45.
14Einleitung
1.5.3 CRP: Regulation
CRP wird überwiegend in der Leber synthetisiert. Die Synthese von CRP wird vor allem auf
Transkriptionsebene reguliert. Aber auch posttranskriptionelle Mechanismen, wie Regulation
der Freisetzung aus dem endoplasmatischen Retikulum durch eine Carboxylesterase, spielen
eine wichtige Rolle46. Die Synthese ist abhängig von Zytokinen, insbesondere von Interleukin
6 (IL-6), Interleukin 1 (IL-1) und Tumornekrosefaktor alpha (TNF-α). Diese bewirken eine
Steigerung der CRP - Produktion, wobei IL-1, Glukokortikoide und Komplementprodukte die
Wirkung von IL-6 zusätzlich verstärken. Die Quelle dieser Zytokine sind Monozyten und
Makrophagen, die direkt am Ort des entzündlichen Geschehens sind. Zusätzlich zur
hepatischen Synthese gibt es allerdings auch Hinweise für eine extrahepatische Produktion in
Lymphozyten47;48, in Makrophagen42, im Pankreasgewebe49 im respiratorischen Epithel43 und
auch im arteriosklerotischen Plaque selbst50.
1.5.4 CRP: Liganden
Phosphatidylcholin (PCh) ist der wichtigste CRP-Bindungspartner (Abb. 8). Es ist Bestandteil
von membrangebundenen Kohlenhydraten, Lipopolysacchariden und Teichonsäuren von
Bakterien, Pilzen, Pflanzen und anderen Mikroorganismen. Die Bindung von CRP an PCh
erfolgt calciumabhängig. Phosphatidylcholin ist in fast allen biologischen Zellmembranen
vorhanden und bildet hier die polare Gruppe von Lecithin und Sphingomyelin. CRP kann hier
in Anwesenheit von Lysolecithin binden. Lysolecithin entsteht, wenn die Zellmembran
beschädigt wird und daraufhin Bestandteile der inneren Membran nach außen gelangen und
mit Phospholipase A2, einem in der Leber produzierten Akut-Phase-Protein, reagieren. So
kann CRP nicht in gesundem und nur in nekrotischem und entzündetem Gewebe binden.
Die weite Verbreitung von Phosphatidylcholin in Polysaccariden in zellulären Membranen
erlaubt CRP eine Reihe von pathogenen Mikroorganismen sowie die Membranen von
beschädigten und nekrotischen Zellen zu erkennen. Gebundenes CRP kann das
Komplementsystem auf klassischem Weg über C1q, das Erkennungsprotein für die
Aktivierung des klassischen Komplementweges aktivieren. Mehrere gebundene CRP-
Moleküle sind nötig, um C1q zu binden und die C3-Konvertase zu aktivieren. Den
alternativen Weg des Komplementsystems kann CRP über Faktor H blockieren31. Hier kommt
es nur zur Bildung des C3-Konvertase-Komplex. Dadurch wird C3 und C4 zur Opsonisierung
gebildet und die Bildung des terminalen Membranangriffskomplexes bleibt aus. Außerdem
15Einleitung
kann CRP an LDL (siehe Kapitel 1.5.5 CRP: Interaktion mit LDL) und Fcγ-Rezeptoren
(siehe Kapitel 1.7 CRP-Rezeptoren) binden.
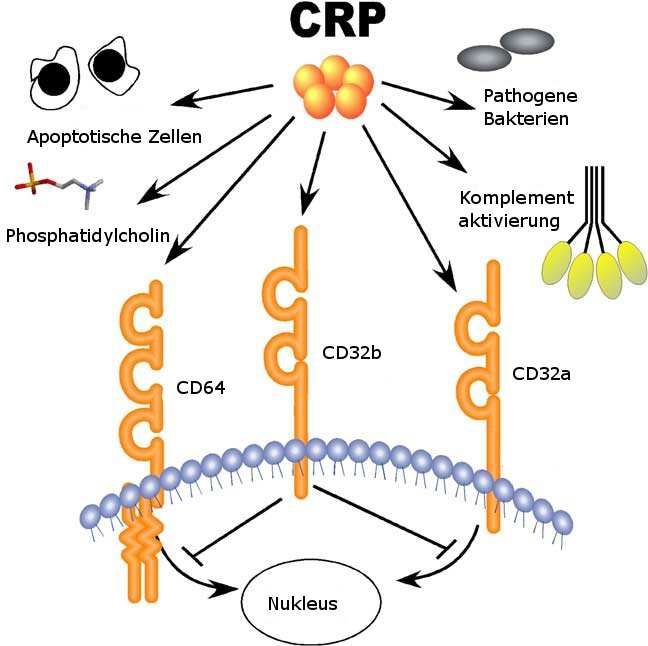
Abb. 8 CRP Liganden. Der wichtigste Ligand für CRP ist Phosphatidylcholin, das auf vielen
Bakterienwänden und beschädigten Zellmembranen vorkommt. Die Bindung von CRP aktiviert
Komplement und opsonisiert sie für die Phagozytose. CRP bindet auch an CD64 und CD32 auf
der Oberfläche von Leukozyten und anderen Zellen. CD64 und CD32a sind aktivierende
Rezeptoren. Aktivierung der Rezeptoren erhöht die Phagozytose und schüttet inflammatorische
Zytokine aus. CD32b ist ein inhibitorischer Rezeptor und die Aktivierung des Rezeptores
blockiert ein aktivierendes Signal.
Aus Marnell et al51
16Einleitung
1.5.5 CRP: Interaktion mit LDL
Verschiedene Arbeiten konnten zeigen, dass CRP sowohl natives LDL52,53 mit dem darin
enthaltene Phosphatidycholin54, als auch oxLDL und das darin enthaltene oxidierte
Phosphatidylcholin binden kann55. Andere Untersuchungen zeigten, dass CRP vor allem
oxLDL und apoptotische Zellen über einen Phosphatidylcholinteil binden kann, der durch
Oxidation des Phosphatidylcholinteils zugänglich wird56,57.
1.6 CRP und Arteriosklerose
Für erhöhte Spiegel von CRP im Blut konnte in zahlreichen Studien ein erhöhtes Risiko für
kardiovaskuläre Ereignisse und anderen Folgeerkrankungen der Arteriosklerose nachgewiesen
werden. Die erste Arbeit dazu stammte von de Beer et al58 aus dem Jahr 1982. Gemessen
wurden CRP und CK-MB (creatin kinase muscle brain), sowohl bei Patienten mit
kardiovaskulären Erkrankungen als auch bei einer Kontrollgruppe. Es zeigte sich ein Anstieg
des CRPs bei Patienten mit Myokardinfarkt, der mit dem Anstieg der CK-MB korrelierte. In
einer Studie an 27939 Frauen konnten Ridker et al zeigen, dass erhöhtes CRP neben den
klassischen Risikofaktoren aus der Framinghamkohorte ein weitere Risikofaktor für
kardiovaskuläre Erkrankungen ist59. Ähnliche Ergebnisse lieferten weiteren Studien5,7.
Haverkate et al konnten in einer Studie mit 2121 Patienten zeigen, dass erhöhte CRP-
Blutspiegel einen prädiktiven Wert für koronare Ereignisse in Patienten mit stabiler oder
instabiler Angina pectoris haben6. Erhöhte CRP-Spiegel bereits bei Krankenhausaufnahme
bedeuten bei Patienten mit instabiler Angina pectoris eine schlechtere Prognose60.
Arteriosklerose mit ihren Folgeerkrankungen stellt bei Dialysepatienten eine der häufigsten
Komplikationen und Todesursachen dar. Interessanterweise sind bei Dialysepatienten die
CRP-Spiegel im Mittel 5-10x höher als bei gesunden Kontrollpersonen. Dies korrelierte mit
einer erhöhten kardiovaskulären- und Gesamtmortalität61,62. Tataru et al untersuchten in ihrer
Studie, in welchem Zusammenhang Plasma-CRP und der Grad der Arteriosklerose stehen. Zu
diesem Zweck wurde bei 1411 Myokardinfarktpatienten der Stenosegrad der großen Becken-
und Beingefäße sowie der extrakraniellen Kopfgefäße mittels Ultraschall untersucht. Es zeigt
sich ein direkter Zusammenhang zwischen der Serum-CRP-Konzentration und dem Grad der
Arteriosklerose: Je höher die CRP-Konzentration, desto ausgeprägter die Arteriosklerose63.
Auch der angiographisch festgestellte Grad der Arteriosklerose war mit dem CRP-Spiegel
17Einleitung
assoziiert. Im Hasen konnte eine Korrelation zwischen der Plaquegröße und den CRP-
Spiegeln nachgewiesen werden. Das CRP konnte hier auch in den Plaques direkt
nachgewiesen werden64. Der CRP-Spiegel hat auch einen prädiktiven Wert für andere
arteriosklerotische Folgekrankheiten wie Schlaganfall65, periphere arterielle Verschlüsse66
und plötzlichen Herztod67. Einige Studien kamen zu weniger deutlichen Ergebnissen; so
untersuchten Danesh et al in der Reykjavikstudie mit 18569 Teilnehmern den Einfluss von
CRP als prädiktiven Wert für Koronare Herzkrankheit. Dort zeigte CRP nur eine moderate
Vorhersagekraft für die koronare Herzkrankheit68.
Die Einführung des sogenannten „high-sensitivity CRP“ (hs-CRP) assays ermöglichte die
Messung von CRP-Konzentrationen im niedrigen Bereich. Ein hs-CRP Spiegel niedriger als
1.0 mg/L wird als niedriges Risiko für die Entwicklung von kardiovaskulären Erkrankungen
eingestuft, zwischen 1.0 und 3.0 mg/L als mittleres Risiko und ein CRP-Spiegel höher als
3.0mg/L als hohes Risiko69. Besonders Patientengruppen mit niedrigem LDL-Spiegel und
daher sonst niedrigem Risiko, die aber einen hohen CRP-Spiegel haben, konnten hier als
Risikogruppe identifiziert werden70.
Es gibt inzwischen auch zahlreiche Hinweise darauf, dass CRP nicht nur für die
Prognoseeinschätzung Wert hat, sondern auch selbst ursächlich an der Entstehung von
Arteriosklerose und ihren Folgekrankheiten beteiligt ist. So untersuchten Danenberg et al in
mit menschlichem CRP transgenen Mäusen nach einer endothelialer Verletzung die Bildung
von Thrombosen71. Dort zeigten sich bei den transgenen Tieren frühere und häufigere
thrombotische Verschlüsse. Relevant ist dies, da das endogene CRP der Maus kein Akut-
phase-Protein ist und auch nur in Spuren synthetisiert wird72. In ApoE-Knockoutmäusen,
einem Tiermodell für Arteriosklerose, war bei mit menschlichem CRP transgenen Tieren das
Fortschreiten der Arteriosklerose beschleunigt und CRP konnte in den Läsionen
nachgewiesen werden73.
18Einleitung
1.6.1 Die CRP-Konfigurationen in der Arteriosklerose
Die beiden CRP-Konfigurationen haben im Stoffwechsel der Endothelzellen zahlreiche
Effekte. Sie führen zur Ausschüttung von Zytokinen und Chemokinen und beeinflussen die
Bildung von Adhäsionsproteinen, die Aggregation von neutrophilen Granulozyten, die
Bildung von Sauerstoffradikalen und die Funktion des Komplementsystems (siehe Einleitung
Kapitel 1.3). Die Effekte auf den Stoffwechsel der Endothelzellen unterscheiden sich
zwischen den beiden CRP-Konfigurationen. Natives CRP hat zahlreiche pro- und
antiinflammatorische Effekte im Organismus, wobei die antiinflammatorischen Effekte, wie
eine Verminderung der Chemotaxis, der Adhäsion an Endothelzellen und der Migration von
neutrophilen Granulozyten zu überwiegen scheinen74. In nCRP behandelten ApoE-Mäusen
war die Endothelabhängige Gefäßrelaxation eingeschränkt75. In menschlichen Endothelzellen
konnten Devaraj et al proatherogene Effekte durch nCRP in der Beeinflußung von IL-8, PAI-
1 und eNOS feststellen76. M-CRP hingegen hat eher proinflammatorische Effekte74, wie unter
zahlreichen anderen eine Erhöhung des inflammtorischen Zytokins Interleukin 877 oder eine
verminderte Apoptose in neutrophilen Granulozyten78.
Gegensätzliche Effekte der CRP-Konfigurationen konnten auch in vivo nachgewiesen
werden. In der Arbeitsgruppe Schwedler et al konnte gezeigt werden, dass in ApoE-
Knockoutmäusen, einem Hypercholesterinämiemodell mit beschleunigter Arteriosklerose,
mCRP die Plaquebildung in frühen Stadien verhindern konnte. N-CRP hingegen verstärkte
die Plaquebildung und das Fortschreiten der Arteriosklerose79.
19Einleitung
1.7 CRP-Rezeptoren
Fc-Rezeptoren existieren für alle Klassen von Immunglobulinen, jene für IgG werden Fcγ-
Rezeptoren genannt. Sie sind Mitglieder der Immunorezeptorenklasse der Tyrosinkinasen und
sind auf allen Zellen des Immunsystems vorhanden. In ihrer Aminosäuresequenz verfügen sie
über eine tyrosinhaltige Aktivierungssequenz (Immunoreceptor tyrosin-based activation
motif, ITAM). Fc-Rezeptoren sind wichtig für die Vermittlung antikörpervermittelter
zellulärer Zytotoxizität und Phagozytose, die Produktion von radikalen Sauerstoffspezies und
für die Immunregulation. Die Aktivierung durch Immunkomplexe führt zur Sekretion von
verschiedenen entzündlichen Mediatoren, Zytokinen und Lymphokinen. Die Fcγ-Rezeptoren
nehmen so an der Entstehung und dem Fortschreiten von Entzündungsprozessen teil.
Es werden drei Typen von Fcγ-Rezeptoren unterschieden.Von jeder Fcγ-Rezeptorenklasse
wiederum gibt es mehrere Subtypen (a,b,c). FcγRI (CD64) ist ein Rezeptor mit hoher Affinität
(70kDa), der auf Monozyten, Makrophagen und aktivierten neutrophilen Granulozyten
nachgewiesen wurde. CD32 ist ein niedrig affiner Rezeptor (40kDa) für monomeres IgG, der
auf Monozyten, Makrophagen, Neutrophilen Granulozyten, B-Zellen, Thrombozyten, Epithel
und Endothel nachgewiesen wurde. CD32b ist ein inhibitorischer Rezeptor. FcγRIII (CD16)
ist auch ein niedrig affiner Rezeptor (50-70kDa) für monomeres IgG der auf neutrophilen
Granulozyten, natürlichen Killerzellen, eosinophilen Granulozyten und Makrophagen
nachgewiesen wurde. CD16a wird auf natürlichen Killerzellen und Makrophagen exprimiert
und hat einen Polypeptidmembrananker, wohingegen CD16b, welches auf neutrophilen
Granulozyten exprimiert wird, ein Glykosyl-phosphatidylinositol (GPI) verankertes Protein
ist80. Insgesamt werden die Fcγ-Rezeptoren von 8 Genen codiert: Drei Gene für den
Hochaffinitätsrezeptor CD64 (a,b,c) und fünf Gene für die Rezeptoren mit niedriger Affinität:
CD32(a,b,c) und CD16 (a,b)81 CD32a, -b und -c entstehen durch alternatives Spleißen und
codieren insgesamt sechs Transkripte82,83. Verschiedene Studien zeigten, dass an der
Aufnahme von CRP durch Leukozyten und Monozyten drei Typen von Fcγ-Rezeptoren
beteiligt sind, der Hochaffinitätsrezeptor CD64 und 2 niedrig affine Rezeptoren, CD16 und
CD3251.
Die Bindung von CRP (oder IgG über ihre Fc-Teile) an Fcγ-Rezeptoren löst eine komplexe
Signalkaskade aus, die hier in den wichtigsten Teilen kurz dargestellt ist51: Bekannt wurden
diese Effekte bei Bindung von IgG, aber die durch CRP ausgelösten Effekte dürften sehr
20Einleitung
ähnlich sein. Nach Bindung des Liganden an den Rezeptor auf der Zelloberfläche erfolgt die
Phosphorylierung von Tyrosinresten in der ITAM. Dies führt über verschiedene Kinasen zur
Aktivierung des Transkriptionsfaktor p38. Zusätzlich wird die Phospholipase Cγ2 (PL Cγ2)
aktiviert, die Calcium aus dem endoplasmatischen Retikulum mobilisiert. Die Bindung des
Liganden löst auch eine Aktivierung eines ERK-Kinasen abhängigen Signalweges aus.
Alle diese Signalwege führen letztendlich zur Aktivierung von Proteinen, die Aktin, Myosin,
Membranfusionen (unter anderem für die Aufnahme von Stoffen in die Zelle) und die
Produktion von reaktiven Sauerstoffspezies regulieren84. Dies stellen Teile entzündlicher
Prozesse dar, dessen Unterstützung dann auch durch CRP über diese Fcγ-Rezeptoren erklärt
werden kann.
1.7.1 CD16 (FcγRIII)
Effekte von mCRP können durch Anti-CD16 Antikörper blockiert werden. In einer
Untersuchung von Khreiss et al konnte gezeigt werden, dass die proinflammatorischen Effekte
von mCRP, die gesteigerte Produktion von verschiedenen Zytokinen in menschlichen
Koronararterienendothelzellen und Leukozyten, durch einen CD16 Antikörper reduziert
werden konnten85,86. M-CRP, nicht aber nCRP, wird durch einen Anti-CD16-Antikörper
daran gehindert an neutrophile Granulozyten zu binden, was darauf hindeutet, dass CD16 der
Rezeptor für mCRP auf neutrophilen Granulozyten ist77.
1.7.2 CD32 (FcγRII)
CRP scheint seine biologischen Effekte vor allem durch CD32 zu vermitteln. Devaraj et al
konnten zeigen, dass in menschlichen Aortenendothelzellen biotinilisiertes CRP (bCRP)
durch Fcγ-Rezeptoren gebunden und aufgenommen wird und die biologischen Effekte des
CRPs vermittelt. So regulierte CRP CD32 hoch, und die Inkubation mit Antikörpern gegen
CD32 reduzierte die Bindung an die Zellen. In der Konfokalmikroskopie wurde CRP
zusammen mit CD32 nachgewiesen. Die Erhöhung von IL-8, ICAM-1, VCAM-1 und die
Erniedrigung der eNOS und des Prostazyklins durch nCRP konnte durch Antikörper gegen
CD32 verhindert werden87. Dieselbe Arbeitsgruppe konnte später Ähnliches mit nCRP in
menschlichen Aortenendothelzellen zeigen. Dort regulierte nCRP die NF-κB-Aktivität hoch,
was zu einer Erhöhung von ICAM und VCAM und der Adhäsion von Monozyten an das
21Einleitung
Endothel führte. Anti-CD32 Antikörper konnten dies verhindern88. CRP induziert in HUVEC
auch die Expression von CD3289. In Makrophagen kann CRP zusammen mit LDL über CD32
aufgenommen werden90. Zusammengefasst deutet dies darauf hin, dass CD32 auf vielen
Zellen der Rezeptor für nCRP ist.
1.7.3 CD64 (FcγRI)
CD64 ist als Rezeptor für CRP auf Leukozyten und Monozyten bekannt51. Für eine
Beteiligung an der Arteriosklerose fanden sich jedoch bislang keine Hinweise, so dass im
Rahmen dieser Arbeit die Untersuchungen auf CD16 und CD32 beschränkt wurden.
1.7.4 Zusammenfassung
Insgesamt also gibt es deutliche Hinweise dafür, dass die Fcγ-Rezeptoren die Rezeptoren für
CRP auf Leukozyten und Monozyten sind, wohingegen CD32 der Rezeptor für nCRP zu sein
scheint, und CD16 der Rezeptor für mCRP. Für eine Rolle als Rezeptoren auch auf
Endothelzellen liegen Hinweise vor. N-CRP-Effekte (wie IL-Induktion, Erniedrigung eNOS
und Prostacyclin) können durch Anti-CD32-Antikörper86-90 aufgehoben werden, mCRP-
Effekte (NO-Bildung, Induktion IL-8, ICAM-1, E-Selectin, VCAM-1) können durch Anti-
CD16 Antikörper- blockiert werden85,86.
Tebo et al fanden zudem Hinweise auf die Existenz zumindest eines weiteren unbekannten
CRP-Rezeptors, CRP konnte auch noch nach Blockierung von CD16 und CD32 an
Monozyten binden91.
22Einleitung
1.8 Fragestellungen
In einer Vorarbeit der Arbeitsgruppe Schwedler et al wurde gezeigt, dass in ApoE-
Knockoutmäusen nCRP die Plaquebildung und das Fortschreiten der Arteriosklerose verstärkt
wurde, wohingegen mCRP die Plaquebildung in frühen Stadien verhindern konnte79.
Immunhistochemische Untersuchungen wiesen mCRP im Endothel und in verschiedenen
Gewebearten nach41-43. Im Rahmen der vorliegenden Arbeit untersuchten wir die Aufnahme
von dil-markiertem acLDL im FACS in humanen Endothelzellen. Es wurde die native
Aufnahme von dil-acLDL untersucht, und unter Inkubation der Endothelzellen mit mCRP
oder oxLDL sowie einer Kombination von beiden. Es wurden verschiedenen
Inkubationszeiten (8 Stunden und 24 Stunden) und CRP-Konzentrationen (10 und 100µg/ml)
verwendet. Um auszuschließen dass festgestellte Effekte durch Kontaminaton mit LPS
entstanden waren, wurden Kontrollzellen mit LPS inkubiert. Um die Beteiligung der nach
Literaturdaten vermuteten Rezeptoren CD16 und CD32 an der in den Versuchen festgestellten
verminderten acLDL-Aufnahme nach Inkubation mit mCRP zu bestimmen führten wir
weitergehende Untersuchungen durch. Dazu wurden die Zellen mit Antikörpern gegen
CD16/32 inkubiert und dann die Aufnahme an acLDL gemessen. Als zusätzliche Methode
wurde in der Immunfluoreszenzmikroskopie m/n-CRP visualisiert und die dil-acLDL-
Aufnahme in den Vesikeln in den Endothelzellen lokalisiert. Desweiteren führten wir
Genexpressionsstudien für CD16/32 mit Endothelzellen nach Inkubation mit mCRP, oxLDL
sowie einer Kombination der beiden durch.
23Material und Methoden
2. Material und Methoden
2.1. Chemikalien und Reagenzien
Substanz Bezugsquelle
β-Mercaptoethanol Sigma-Aldrich (München)
BSA (Bovine serum albumin) Sigma-Aldrich (München)
BBE (Bovine brain extract) Cambrex (Verviers, Belgien)
CRP SeraCare Inc. (West Bridgewater, USA)
Diliertes, acetyliertes LDL eigenes Labor
EBM-1 Basismedium (C3121) Cambrex (Verviers, Belgien)
DAPI Roche (Mannheim)
DiL Molecular Probes (Leiden, Niederlande)
Ethanol Baker JT (Griesheim)
FCS (Fetal calf serum) Cambrex (Verviers, Belgien)
FITC (fluorescein isothiocyanat) Sigma-Aldrich (München)
GA-1000 (Amphotericin B, Gentamycin) Cambrex (Verviers, Belgien)
HUVEC Cambrex (Verviers, Belgien)
Hydrocortison Cambrex (Verviers, Belgien)
Lipopolysaccarid (LPS) L6529 Sigma-Aldrich (München)
Modifiziertes CRP Immtech Pharmaceuticals (Vernon Hills, USA)
Natives CRP Immtech Pharmaceuticals (Vernon Hills, USA)
Natriumacetat Merck (Darmstadt)
Oxidiertes LDL eigenes Labor
Paraformaldehyd (4%) Merck (Darmstadt)
Dulbecco’s PBS (ohne Ca2+ und Mg2+) PAA (Linz, Österreich)
Dulbecco’s PBS (mit Ca2+ und Mg2+) Sigma-Aldrich (München)
rhEGF Cambrex (Verviers, Belgien)
Rhodamin Sigma-Aldrich (München)
RLT-Puffer Qiagen (Venlo, Niederlande)
RNAase OFF AppliChem (Darmstadt)
Triton (0,5%) X-100 Sigma-Aldrich (München)
Trypanblau Sigma-Aldrich (München)
24Material und Methoden
Trypsin/EDTA PAA (Linz, Österreich)
Wärmebadzusatz Aquaclean Hartenstein (Würzburg)
2.1.1 Antikörper
Tab. 1 Antikörper Typ Bezugsquelle
CD-16 Antikörper (555404) Monoklonal BD Pharmingen (Heidelberg)
CD-32 Antikörper (555447) Monoklonal BD Pharmingen (Heidelberg)
mCRP-Antikörper (9C9) Monoklonal Immtech Pharmaceuticals
(Vernon Hills, USA)
mCRP-Antikörper (7A8) Monoklonal Immtech Pharmaceuticals
(Vernon Hills, USA)
nCRP-Antikörper (2C10) Monoklonal Immtech Pharmaceuticals
(Vernon Hills, USA)
nCRP-Antikörper (1D6) Monoklonal Immtech Pharmaceuticals
(Vernon Hills, USA)
Maus-Antikörper (Cy2) Polyklonal Dianova (Hamburg)
2.1.2 CRP
Modifiziertes und natives CRP wurden von Immtech. Pharmaceuticals Inc. (Vernon Hills,
Illinois) bezogen und auf folgende Weise hergestellt:
2.1.2.1 Natives CRP (nCRP)
Das CRP wurde von SeraCare Inc. erstanden, an Q-Sepharose FF gebunden und mit einem
schrittweisen NaCl-Gradienten gelöst und gesammelt. Der Ionenaustausch mit CRP ist
wichtig, um eventuell anfallendes Endotoxin zu minimieren und auch um jegliches mCRP,
das sich dort befinden könnte, zu entfernen (Es würde an die Agarose binden). Anschließend
wurde sofort CaCl2 hinzugefügt (2 mmol/L). Das nCRP wurde dann gegen 25 mmol/L-Tris-
25Material und Methoden
HCl und 0.15 mol/L NaCl mit 2 mmol/L CaCl2 dialysiert, steril filtriert und bei 4°C
aufbewahrt. Die Aufbewahrung in Calcium-enthaltenden Puffern ist notwendig, um die
spontane Formation von mCRP aus nCRP zu verhindern. Siehe auch Darstellung in
Schwedler et al79.
2.1.2.2 Modifiziertes CRP (mCRP)
M-CRP wurde aus nCRP durch Chelatierung mit Harnstoff hergestellt. Eine rekombinante
Form von mCRP (rmCRP), dessen beide Cysteinreste zu Alaninresten mutiert wurden und das
mit einem angefügten N-terminalen Formylmethioninrest ergänzt wurde, wurde in
Escherichia coli exprimiert und aus Einschlusskörperchen zu >95% Reinheit isoliert. Um die
Reinheit von rmCRP zu erhöhen, wurde es mit Methylmaleinsäureanhydrid acetyliert, um so
die Löslichkeit zu erhöhen (C-rmCRP). Die Aminosäurenzusammensetzung und N-terminale
Sequenz von C-rm CRP und mCRP waren, wie in der Polyacrylamid-Gelelektrophorese (SDS-
Page) gezeigt werden konnte, direkt vergleichbar. Nach der Aufreinigung wurden die
Methylmaleinsäureanhydridgruppen durch Dialyse entfernt (bei Raumtemperatur für 20
Stunden in 0.1 mol/L Citrat, anschließend weitere Dialyse bei 4°C gegen 25 mmol/L
Natrium-PBS ohne Calcium). Dies ergab ein unlösliches, suspendiertes Protein ähnlich wie
mCRP in salzbasierten Puffern. Monoklonale Antikörper gegen das C-terminale Oktapeptid
(mAb 9C9), das nur in mCRP und nicht in nCRP exprimiert wird, reagieren mit der gleichen
Spezifität und Affinität mit mCRP, C-rmCRP und Cx-rmCRP. Siehe auch Darstellung in
Schwedler et al79.
2.1.3 Lipoproteine
Oxidiertes und dil-markiertes, acetyliertes LDL wurden im Nephrologischen Labor der
Universität Würzburg nach folgender Weise hergestellt92,93. Für die Präparation von LDL
wurde frisches Plasma von Spendern der Medizinischen Universitätsklinik Würzburg
verwendet. Die Dichte wurde eingestellt und Antioxidantien wurden hinzugegeben. Die LDL-
Präparation wurde dann ultrazentrifugiert (50000 U/min, für 8 Stunden bei 5°C) und die
Schicht, die LDL, VLDL und Triacylglycerine enthält abgesaugt. Als nächster Schritt wurde
die Dichte der LDL-Präparation erneut eingestellt, die Flüssigkeit mit Kalium-Phosphat-
26Material und Methoden
Puffer unterschichtet und ultrazentrifugiert (50000U/min für 3 Stunden bei 5°C). Die LDL-
Bande wurde abgesaugt. Anschließend wurden die Blutsalze durch Filtrierung in Millipore-
Filtereinheiten und Zentrifugation (1500 U/min, für 2 Stunden bei 4°C) abgetrennt. Zum
Abschluß der Präparation des LDL erfolgte dann die Dialysierung und sterile Filtration der
Präparation mit Kalium-Phosphatpuffer in einem Dialyseschlauch mit 50 kDa großen Poren.
Die Oxidierung für die Herstellung von oxLDL erfolgte durch Inkubation von
antioxidantienfreiem LDL mit 1µM/L CuSO4 in PBS für 24 Stunden bei Raumtemperatur. Die
Acetylierung von LDL für die Herstellung von dil-acLDL erfolgte durch Zugabe von
Natriumacetat. Dafür wurde pro ml LDL 1 ml Natriumacetat hinzugefügt und anschließend
auf Eis gestellt. In 5-minütigen Abständen wurde dann Essigsäureanhydrid hinzugegeben (1,5
mg pro mg). Die weiter über 30 Minuten gekühlte LDL-Präparation wurd dann durchmischt
und gegen PBS für 12 Stunden bei 4°C dialysiert. Schließlich wurden 300 µg DiL (1,1’-
Dioctadecyl-1-3,3,3’,3’tetramethyl-indocarbocyanin-perchlortat) in 10µl DMSO gelöst und
zum acLDL hinzugegeben und für 18 Stunden bei 37°C inkubiert (Labelling). Das nun
gelabelte acLDL wurde durch Ultrazentrifugation isoliert, gegen PBS dialysiert und
anschließend steril filtriert. Die Homogenität der Lipoproteine wurde durch Agarosegel-
elektrophorese überprüft. Die Mobilität von oxLDL in der Agarosegel-elektrophorese als
Index für die Lipoproteinoxidation bzw. Acetylierung war bei den verwendeten Präparationen
verglichen mit nativem LDL 2.5-3-fach erhöht. Die Proteinkonzentration wurde mit einem Kit
(Sigma protein kit), das auf einer Methode von Lowry et al basiert, gemessen94. Die
Lipoproteine wurden bei 4°C im Dunkeln aufbewahrt und regelmäßig für die Versuche frisch
hergestellt. Insgesamt wurden 3 verschiedene LDL-Präparationen verwendet.
27Material und Methoden
2.1.4 Endothelzellmodell: HUVEC
Um den Einfluss der verschiedenen CRP-Konfigurationen im LDL-Metabolismus zu
untersuchen, wurden menschlichen Nabenschnurvenenendothelzellen gewählt (Human
Umbilical Vein Endothelial Cells, HUVEC), die von Cambrex bezogen wurden und nach
Anleitung des Herstellers in den folgenden Medien kultiviert wurden.
Zu einem EBM-1 Basismedium für Endothelzellen wurden folgende Zusätze hinzugefügt:
Humaner epidermaler Wachstumsfaktor (rhGEF) 0,1%, Hydrocortison 0,1%, GA-1000
(Amphotericin B und Gentamycin) 0,1%, BBE (bovine brain extract) 0,4%, BSA (bovine
serum albumin) 0,1%. Dieses Hungermedium wurde für die Versuche verwendet, um den
Einfluss der im FCS (fetal calf serum) enthaltenen Stoffe zu vermeiden und die Zellen auf
niedrigem Stoffwechselniveau zu halten und so den Einfluss der CRP-Konfigurationen
möglicherweise besser darstellen zu können. Um HUVEC anzuzüchten, wurde
Wachstumsmedium verwendet. Es enthält die gleiche Zusammensetzung wie das
Hungermedium, nur das 0,1 %-ige BSA wurde durch 10% FCS ersetzt.
Abb. 9 Konfluente HUVEC-Kultur unter dem Mikroskop, 100X
28Sie können auch lesen

















































