Max Delbrück Center for Molecular Medicine
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

https://www.mdc-berlin.de/de/veroeffentlichungstypen/clinical- journal-club Als gemeinsame Einrichtung von MDC und Charité fördert das Experimental and Clinical Research Center die Zusammenarbeit zwischen Grundlagenwissenschaftlern und klinischen Forschern. Hier werden neue Ansätze für Diagnose, Prävention und Therapie von Herz-Kreislauf- und Stoffwechselerkrankungen, Krebs sowie neurologischen Erkrankungen entwickelt und zeitnah am Patienten eingesetzt. Sie sind eingelanden, um uns beizutreten. Bewerben Sie sich!
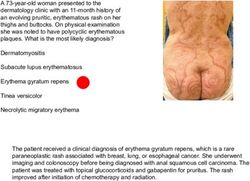
A previously healthy 36-year-old woman presented to the infectious diseases clinic with a 3-day history of fever, arthralgias, myalgias, and headache. She denied any recent travel or new sexual partners but admitted to having a pet rat. Physical examination revealed swollen and tender joints as well as a maculopapular rash on the feet and hands, with pustule formation. What is the most likely diagnosis? The plague Rat bite fever Leptospirosis Tularemia Hantavirus Correct! The patient had been bitten by her pet rat and presented with the findings above. She was admitted to the hospital, and empirical treatment with intravenous ceftriaxone was initiated. Cultures of a blood sample obtained at presentation were positive within 24 hours for Streptobacillus moniliformis, a gram-negative bacillus commonly found in the oropharyngeal flora of rats. The diagnosis of rat bite fever was confirmed. With ongoing treatment, the fever and arthralgias resolved within 3 days and 6 days, respectively. The patient was discharged on hospital day 7 and completed a course of treatment with oral amoxicillin. She remained well at follow-up 3 months later.
Als Schwarzer Tod wird eine der verheerendsten Pandemien der Weltgeschichte bezeichnet, die in Europa zwischen 1346 und 1353 geschätzte 25 Millionen Todesopfer – ein Drittel der damaligen Bevölkerung – forderte. Als Ursache gilt die durch das Bakterium Yersinia pestis hervorgerufene Pest. Seit der Entdeckung des Bakteriums Yersinia pestis gegen Ende des 19. Jahrhunderts war herrschende Meinung, dass es als Erreger für die als Schwarzer Tod bekannte Pandemie verantwortlich sei. Dafür sprechen die Eigenschaften von Yersinia pestis, zu denen ein extrem hohes Ansteckungspotential gehört, die mit der Infektion verbundenen Symptome sowie der Nachweis von Yersinia-DNA in Zahnmark bzw. Skelett von Menschen des 8. und des 14. Jahrhunderts. Alexandre Émile Jean Yersin, (* 22. September 1863 in Lavaux in der Nähe von Aubonne, Schweiz; † 28. Februar 1943 in Nha Trang, Annam, heute Vietnam).
Rat-bite fever is an acute, febrile human illness caused by bacteria transmitted by rodents, in most cases, which is passed from rodent to human by the rodent's urine or mucous secretions. Alternative names for rat-bite fever include streptobacillary fever, streptobacillosis, spirillary fever, bogger, and epidemic arthritic erythema. It is a rare disease spread by infected rodents and can be caused by two specific types of bacteria. Most cases occur in Japan, but specific strains of the disease are present in the United States, Europe, Australia, and Africa. Some cases are diagnosed after patients were exposed to the urine or bodily secretions of an infected animal. These secretions can come from the mouth, nose, or eyes of the rodent. The majority of cases are due to the animal's bite. It can also be transmitted through food or water contaminated with rat feces or urine. Other animals can be infected with this disease, including weasels, gerbils, and squirrels. Household pets such as dogs or cats exposed to these animals can also carry the disease and infect humans. If a person is bitten by a rodent, it is important to quickly wash and cleanse the wound area thoroughly with antiseptic solution to reduce the risk of infection. This condition is diagnosed by detecting the bacteria in skin, blood, joint fluid, or lymph nodes. Blood antibody tests may also be used. To get a proper diagnosis for rat-bite fever, different tests are run depending on the symptoms being experienced. To diagnosis streptobacillary rat-bite fever, blood or joint fluid is extracted and the organisms living in it are cultured. Diagnosis for spirillary rat bite fever is by direct visualization or culture of spirilla from blood smears or tissue from lesions or lymph nodes.

Eine Leptospirose ist eine Infektionskrankheit, die durch bestimmte Krankheitserreger der Gattung Leptospira (aus der Ordnung der Spirochäten) verursacht wird. Beim Menschen wird die Krankheit durch Leptospira interrogans verursacht. Es handelt sich dabei um eine meldepflichtige Zoonose, deren natürliche Wirte vor allem Ratten und Mäuse, im Falle der Schweinehüterkrankheit auch Schweine und Rinder sind. Die Übertragung auf den Menschen erfolgt durch Kontakt mit Urin, Blut oder Gewebe infizierter Tiere bzw. verunreinigtem Wasser, vor allem aus Bächen, Sümpfen, Tümpeln und der Kanalisation. Leptospiren gelangen über den Urin infizierter Säugetiere (Ratten, Hunde, Mäuse, Igel) in die Umwelt. Durch kleine Hautverletzungen oder über die Schleimhaut kann der Mensch sich mit dem Erreger anstecken.
Tularämie ist eine häufig tödlich verlaufende ansteckende Erkrankung bei frei lebenden Nagetieren und Hasenartigen, die durch das Bakterium Francisella tularensis ausgelöst wird. Die Erkrankung kann auf den Menschen übertragen (Zoonose) werden und zählt in Deutschland zu den meldepflichtigen Tierkrankheiten. Da das Beschwerdebild dem der Pest ähnelt und die Erkrankung sehr häufig Hasen und Wildkaninchen befällt, wird sie häufig auch als Hasenpest bezeichnet. Andere Namen sind Nagerpest, Lemmingfieber, Parinaudkrankheit und Hirschfliegenfieber. Zwischen 1919 und 1928 beschäftigte sich Edward Francis sehr ausführlich mit der Erkrankung und benannte sie nach dem Ort Tulare in Kalifornien/USA. Der wissenschaftliche Name des Erregers wurde ebenfalls nach ihm benannt. In Europa wurde die Tularämie zum ersten Mal 1931 dokumentiert, und zwar an der Ostseeküste Mittelschwedens. Zwischen 1936 und 1950 gelang den sowjetischen Wissenschaftlern H. A. Gaiski, B. Y. Elbert, Somov und Chatenever die Entwicklung eines Impfstoffes gegen die Tularämie. Der Erreger der Tularämie ist das hochansteckende Bakterium Francisella tularensis (früher auch: Pasteurella tularensis). Es handelt sich um ein sehr kleines, gramnegatives, kokkoides, sporenloses, schwer anzüchtbares Stäbchen, das den γ- Proteobakterien zugeordnet wird.
Die Familie Hantaviridae aus der Ordnung der Bunyavirales umfasst neben wenigen Spezies der Gattungen Loanvirus, Mobatvirus und Thottimvirus vor allem zahlreiche Arten der Gattung Orthohantavirus: unter anderem die humanpathogenen Arten Hantaan-Virus (HTNV), Puumala-Virus (PUUV), Dobrava-Belgrad-Virus (DOBV), Seoul-Virus (SEOV), Sin-Nombre-Virus (SNV) und Andes-Virus (ANDV). Diese behüllten Einzel-Strang(−)-RNA- Viren [ss(−)RNA] verursachen je nach Virustyp verschiedene Erkrankungen. Dazu zählen schwere Lungenerkrankungen (Pneumonie), akutes Nierenversagen (Nephrotisches Syndrom) oder hämorrhagische Fiebererkrankungen. Die Viren sind weltweit verbreitet und treten auch in Mitteleuropa auf. Sie werden durch den Kot oder Urin infizierter Nagetiere (Mäuse und Ratten), der als Staub eingeatmet wird, auf den Menschen übertragen. Die infizierten Nagetiere selbst zeigen keine Krankheitssymptome. Die menschlichen Erkrankungen verlaufen unterschiedlich schwer. Während die in Mitteleuropa auftretenden Puumala- und Dobrava-Virus- Infektionen in weniger als 1 Prozent der klinisch auffälligen Fälle tödlich verlaufen, beträgt die Letalität bei Infektionen mit dem in Ostasien auftretenden Hantaan-Virus und mit dem auf dem Balkan zu findenden Dobrava- Virus bis zu 15 Prozent und bei den amerikanischen Hantaviren (Sin-Nombre-Virus, Andes-Virus und andere) etwa 30–40 Prozent. Der Name Hanta geht auf den Fluss Hantan in Südkorea zurück, an dem in den 1950er- Jahren während des Koreakrieges mehr als 3.000 amerikanische Soldaten an einem ungewöhnlich starken Fieber mit anschließend häufigen Nierenversagen erkrankten.
Die diastolische Herzinsuffizienz, auch Herzinsuffizienz bei erhaltener systolischer Pumpfunktion (HFpEF von engl. heart failure with preserved ejection fraction) oder Herzinsuffizienz mit erhaltener systolischer linksventrikulärer Funktion, ist eine Form der Linksherzinsuffizienz. Wenn die diastolische Herzinsuffizienz zusammen oder als Folge einer arteriellen Hypertonie (Bluthochdruck) auftritt, wird sie auch als Hypertensive Herzkrankheit bezeichnet. Bei der diastolischen Herzinsuffizienz liegt eine Funktionsstörung in der Entspannungsphase des Herzens (Diastole) vor. Die Pumpfunktion des Herzens (Systole) ist nicht oder nur wenig beeinträchtigt. Die asymptomatische diastolische Dysfunktion ist eine Vorstufe der diastolischen Herzinsuffizienz. Zwischen 22 und 73 % aller Patienten, die Symptome einer Herzinsuffizienz aufweisen, leiden an einer isolierten diastolischen Funktionsstörung. ie diastolische Herzinsuffizienz ist definiert als erhöhter Füllungswiderstand vorwiegend der linken Herzkammer bei normaler systolischer Pumpfunktion. Die Herzkammer hat entweder eine eingeschränkte aktive Entspannungsfähigkeit (Relaxation) u. a. infolge von: Durchblutungsstörung (Ischämie), Wanddickenzunahme (Hypertrophie durch Bluthochdruck, Druckbelastung bei Herzklappenerkrankungen, hypertrophe Kardiomyopathie) oder Alterung
Neprilysin ist ein im menschlichen Körper weit verbreitetes Enzym, vor allem in den Nieren und der Lunge. Es ist auch unter den Namen Neutrale Endopeptidase und Membran-Metallo-Endopeptidase (MME) bekannt und wird durch das Gen MME codiert. Darüber hinaus spielt es in der Onkologie und in der Immunhistochemie eine große Rolle, dort unter den synonymen Bezeichnungen CD 10 (Cluster of Differentiation 10) und CALLA (common acute lymphoblastic leukemia antigen). Neprilysin ist eine Zink(II)-abhängige membrangebundene Metalloprotease, wobei der auf der Zellmembran sitzende Teil des Proteins (die Ektodomäne) dann in den Extrazellularraum freigesetzt wird. Dort baut Neprilysin zahlreiche Peptidhormone durch enzymatische Spaltung am Amino-Terminus hydrophober Aminosäuren ab. Hierzu gehören Hormone wie Glukagon, aber auch zahlreiche parakrin wirkende Peptide wie Bradykinin, Oxytozin, Substanz P, Endothelin und natriuretische Peptide wie ANP und BNP, aber auch Beta-Amyloid. Sacubitril, ein Neprilysin- Inhibitor, wurde mit dem AT1-Antagonisten Valsartan kombiniert im Wirkstoff LCZ696 in der PARADIGM-HF- Studie an 8442 Patienten zur Therapie der Herzinsuffizienz eingesetzt. LCZ696 (Markenname Entresto) zeigte in dieser Studie ein besseres Ergebnis als der ACE-Hemmer Enalapril, was eventuell auf die Hemmung des Abbaus der natriuretischen Proteine zurückzuführen ist. Medikamente dieser Gruppe werden als Angiotensin-Rezeptor-Neprilysin-Inhibitoren (ARNI) bezeichnet.

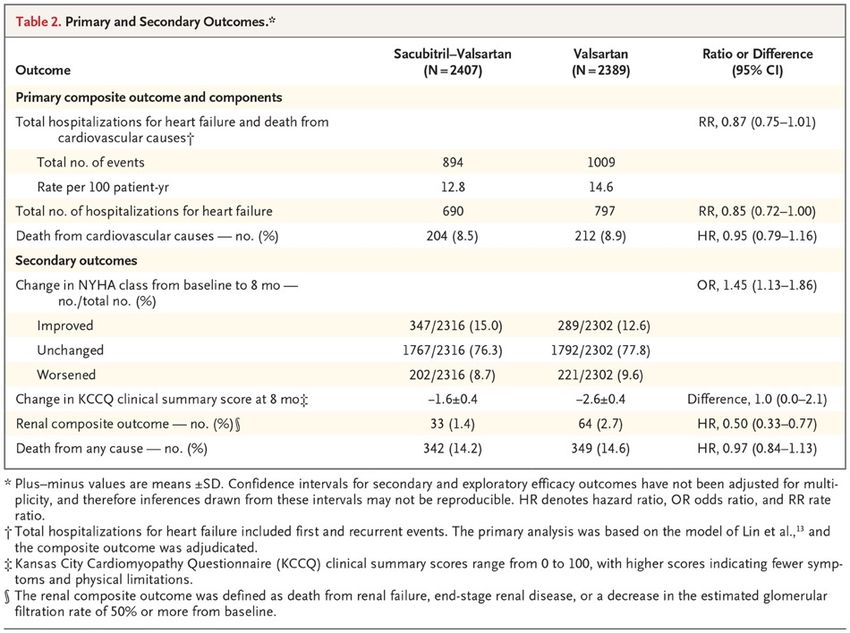
Angiotensin–Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction The angiotensin receptor–neprilysin inhibitor sacubitril– valsartan led to a reduced risk of hospitalization for heart failure or death from cardiovascular causes among patients with heart failure and reduced ejection fraction. The effect of angiotensin receptor–neprilysin inhibition in patients with heart failure with preserved ejection fraction is unclear. We randomly assigned 4822 patients with New York Heart Association (NYHA) class II to IV heart failure, ejection fraction of 45% or higher, elevated level of natriuretic peptides, and structural heart disease to receive sacubitril–valsartan (target dose, 97 mg of sacubitril with 103 mg of valsartan twice daily) or valsartan (target dose, 160 mg twice daily). The primary outcome was a composite of total hospitalizations for heart failure and death from cardiovascular causes. Primary outcome components, secondary outcomes (including NYHA class change, worsening renal function, and change in Kansas City Cardiomyopathy Questionnaire [KCCQ] clinical summary score [scale, 0 to 100, with higher scores indicating fewer symptoms and physical limitations]), and safety were also assessed.
Time-to-Event Curves for Primary Composite Outcome and Its Components. Panel A shows Ghosh–Lin curves for the primary composite outcome of total hospitalizations for heart failure and death from cardiovascular causes, Panel B Ghosh–Lin curves for total hospitalizations for heart failure, and Panel C Kaplan–Meier curves for death from cardiovascular causes. Insets show the same data on an enlarged y axis.
Primary Outcome in Prespecified Subgroups. The primary outcome was a composite of total hospitalizations for heart failure and death from cardiovascular causes. Race was reported by the patient. New York Heart Association (NYHA) class may have changed between screening and randomization. The diamond indicates the overall effect, the size of the boxes is proportional to the number of patients in the subgroup, and arrows indicate that the upper or lower boundary of the confidence interval is off the scale. ACE denotes angiotensin-converting enzyme, GFR glomerular filtration rate, and NT- proBNP N-terminal pro–B-type natriuretic peptide.
Discussion In this trial involving patients with heart failure and preserved left ventricular ejection fraction, we compared treatment with sacubitril–valsartan with treatment with valsartan alone. The primary composite outcome of total hospitalizations for heart failure and death from cardiovascular causes did not differ significantly between the two groups. The findings from nine prespecified supportive and sensitivity analyses were consistent with those from the primary analysis. There were fewer primary outcome events with sacubitril–valsartan than with valsartan, and an analysis of investigator-reported primary outcomes suggested a benefit of this therapy. There was a modest, although statistically nonsignificant, lower rate of hospitalizations for heart failure with sacubitril–valsartan than with valsartan and no significant difference in the risk of death from cardiovascular causes. Of four prespecified secondary outcomes, which were considered to be exploratory, the change in the NYHA class from baseline to month 8 and the occurrence of a decline in renal function favored sacubitril–valsartan over valsartan. Sacubitril–valsartan was associated with a higher incidence of hypotension and angioedema but a lower incidence of elevated serum creatinine or potassium levels than valsartan. We did not find a significant benefit of sacubitril–valsartan in patients with heart failure with preserved ejection fraction with respect to the primary composite outcome of total hospitalizations for heart failure and death from cardiovascular causes. In the context of known benefit of this treatment in patients with heart failure and left ventricular systolic dysfunction, and with the suggestion of a differential effect of sacubitril–valsartan in our trial in relation to left ventricular ejection fraction, future research should focus on the potential role of angiotensin receptor–neprilysin inhibition in patients with heart failure and ejection fraction that is below normal but not frankly reduced.
P2Y12 is a chemoreceptor for adenosine diphosphate (ADP) that belongs to the Gi class of a group of G protein-coupled (GPCR) purinergic receptors. This P2Y receptor family has several receptor subtypes with different pharmacological selectivity, which overlaps in some cases, for various adenosine and uridine nucleotides. The P2Y12 receptor is involved in platelet aggregation and is thus a biological target for the treatment of thromboembolisms and other clotting disorders. Two transcript variants encoding the same isoform have been identified for this gene. In the field of purinergic signaling, the P2Y12 protein is found mainly but not exclusively on the surface of blood platelets, and is an important regulator in blood clotting. The drugs clopidogrel (Plavix), prasugrel (Efient, Effient), ticagrelor (Brilinta), and cangrelor (Kengreal) bind to this receptor and are marketed as antiplatelet agents. A network meta-analysis of 37 studies involving 88,402 STEMI patients and 5,077 major adverse cardiac events (MACE) patients found that use of prasugrel was associated with lower mortality and MACE than other drugs in this class (clopidogrel and ticagrelor).
Cytochrome P450 2C19 (abbreviated CYP2C19) is an enzyme. This protein, a member of the cytochrome P450 mixed-function oxidase system, is involved in the metabolism of xenobiotics, including many proton pump inhibitors and antiepileptics. In humans, the CYP2C19 protein is encoded by the CYP2C19 gene. CYP2C19 is a liver enzyme that acts on at least 10% of drugs in current clinical use, most notably the antiplatelet treatment clopidogrel (Plavix) also drugs that treat pain associated with ulcers, such as omeprazole, antiseizure drugs such as mephenytoin, the antimalarial proguanil, and the anxiolytic diazepam. Genetic polymorphism (mainly CYP2C19*2, CYP2C19*3 and CYP2C19*17) exists for CYP2C19 expression, with approximately 3–5% of European and 15–20% of Asian populations being poor metabolizers with no CYP2C19 function. This may reduce the efficacy of clopidogrel (Plavix). The basis for this reduced effect of clopidogrel in patients who have a gene of reduced activity may seem somewhat paradoxical, but can be understood as follows. Clopidogrel is administered as a “prodrug;” that is, a drug that is inactive when taken, and then depends on the action of an enzyme in the body in order to be activated. In patients who have a gene of reduced activity, clopidogrel may not be metabolized to its active form.
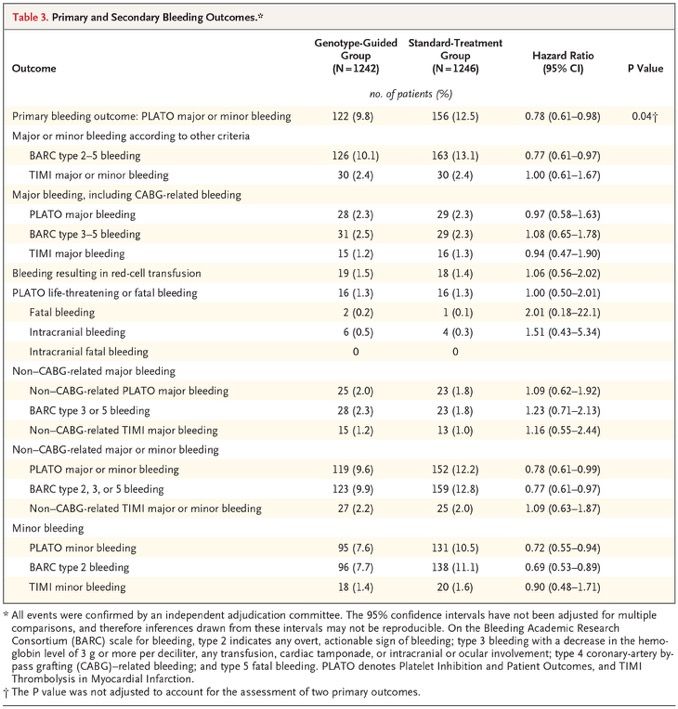
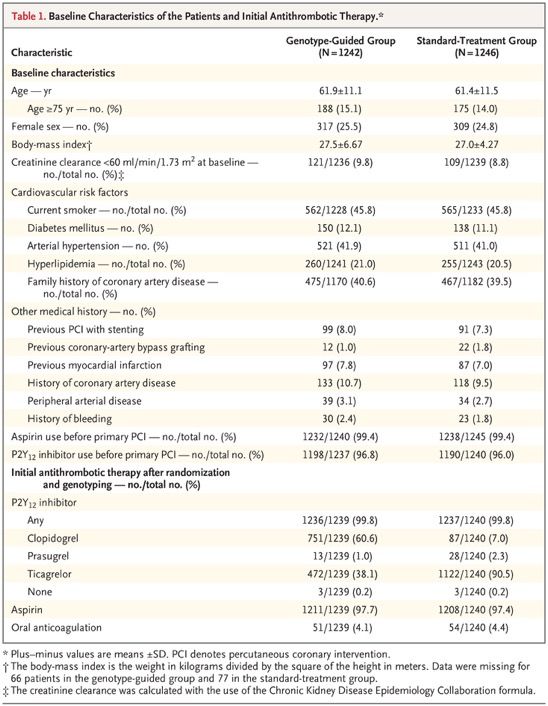
A Genotype-Guided Strategy for Oral P2Y12 Inhibitors in Primary PCI It is unknown whether patients undergoing primary percutaneous coronary intervention (PCI) benefit from genotype-guided selection of oral P2Y12 inhibitors. We conducted a randomized, open-label, assessor-blinded trial in which patients undergoing primary PCI with stent implantation were assigned in a 1:1 ratio to receive either a P2Y12 inhibitor on the basis of early CYP2C19 genetic testing (genotype-guided group) or standard treatment with either ticagrelor or prasugrel (standard-treatment group) for 12 months. In the genotype-guided group, carriers of CYP2C19*2 or CYP2C19*3 loss-of-function alleles received ticagrelor or prasugrel, and noncarriers received clopidogrel. The two primary outcomes were net adverse clinical events — defined as death from any cause, myocardial infarction, definite stent thrombosis, stroke, or major bleeding defined according to Platelet Inhibition and Patient Outcomes (PLATO) criteria — at 12 months (primary combined outcome; tested for noninferiority, with a noninferiority margin of 2 percentage points for the absolute difference) and PLATO major or minor bleeding at 12 months (primary bleeding outcome). The initial plan was for the trial to compare a genotype-guided strategy for selecting oral P2Y12 inhibitors with a standard-treatment strategy for which clopidogrel was recommended. However, the 2011 European Society of Cardiology (ESC) guideline for patients with acute coronary syndrome without ST-segment elevation recommended use of ticagrelor or prasugrel over clopidogrel. In anticipation that the same recommendation would be made for patients with ST-segment elevation, we changed the treatment in the standard-treatment group from clopidogrel to ticagrelor or prasugrel. Because we were now testing guided de-escalation of therapy instead of guided escalation of therapy, there were major changes in the primary outcomes and hypotheses of the trial.
Incidence Curves for the Primary Outcomes. Panel A shows the cumulative incidence of the primary combined thrombotic and bleeding outcome, consisting of death from any cause, myocardial infarction, definite stent thrombosis, stroke, or major bleeding defined according to Platelet Inhibition and Patient Outcomes (PLATO) criteria. Panel B shows the primary bleeding outcome of PLATO major or minor bleeding. The inset in each panel shows the same data on an enlarged y axis. PCI denotes percutaneous coronary intervention.
Discussion In this trial, we investigated the possible clinical benefit of CYP2C19 genotype–guided antiplatelet therapy in patients with STEMI undergoing primary PCI. There are two key findings from this trial. First, the use of a genotype-guided strategy, in which patients without a CYP2C19 loss-of-function allele received clopidogrel, was not associated with a higher risk of combined death from any cause, myocardial infarction, definite stent thrombosis, stroke, or major bleeding 12 months after primary PCI than standard treatment with the more potent P2Y12 inhibitors ticagrelor and prasugrel. Second, the use of clopidogrel in the genotype-guided group resulted in a lower risk of (mostly minor) bleeding than standard treatment. However, the incidence of bleeding was not significantly lower in the de-escalation group than in the prasugrel group. Furthermore, this method of guiding therapy requires patients to switch between P2Y12 inhibitors multiple times if they have high platelet reactivity during treatment with clopidogrel, and it requires patients to revisit the clinic to perform platelet-function testing. On the basis of the TROPICAL-ACS trial, the latest ESC guidelines give a class IIb recommendation to use platelet-function testing for guided de-escalation, especially in patients who are not deemed to be candidates for 12 months of potent antiplatelet therapy. In conclusion, in patients with STEMI undergoing primary PCI, a CYP2C19 genotype–guided strategy for selection of oral P2Y12 inhibitor therapy was noninferior to standard treatment with ticagrelor or prasugrel at 12 months with respect to thrombotic events and resulted in a lower incidence of bleeding. Editorial (Dan Roden) The POPular Genetics trial provides strong support for a genotype-guided approach to clopidogrel prescribing in patients of European ancestry, in whom the contribution of CYP2C19 variants was first defined; a minority of patients of European ancestry carry loss-of-function variants, and very few are poor metabolizers. The result has even greater implications for parts of the world where these variants are much more common. Professional societies, which increasingly view atherosclerosis as a worldwide epidemic, must now rethink their stance with respect to genotyping to improve the effectiveness of clopidogrel therapy.
Lancet review on colon cancer last week
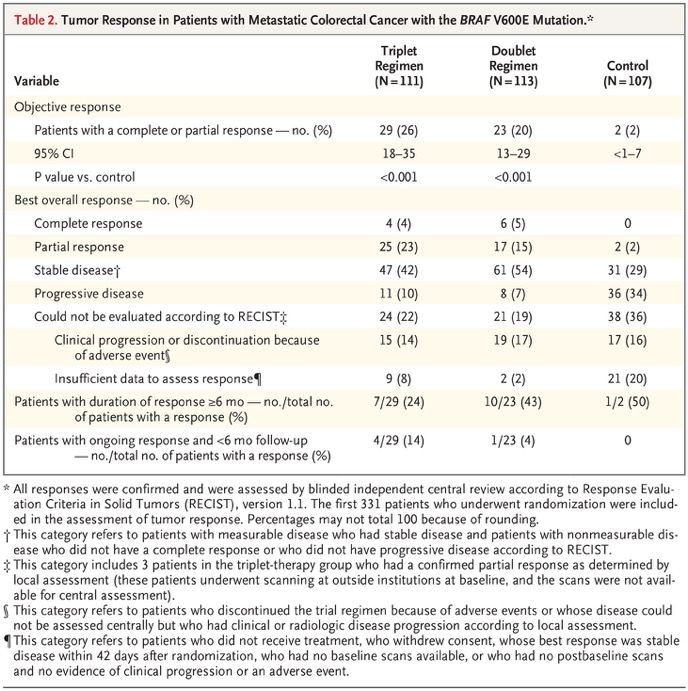
Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer Patients with metastatic colorectal cancer with the BRAF V600E mutation have a poor prognosis, with a median overall survival of 4 to 6 months after failure of initial therapy. Inhibition of BRAF alone has limited activity because of pathway reactivation through epidermal growth factor receptor signaling. In this open- label, phase 3 trial, we enrolled 665 patients with BRAF V600E–mutated metastatic colorectal cancer who had had disease progression after one or two previous regimens. Patients were randomly assigned in a 1:1:1 ratio to receive encorafenib, binimetinib, and cetuximab (triplet-therapy group); encorafenib and cetuximab (doublet-therapy group); or the investigators’ choice of either cetuximab and irinotecan or cetuximab and FOLFIRI (folinic acid, fluorouracil, and irinotecan) (control group). The primary end points were overall survival and objective response rate in the triplet- therapy group as compared with the control group. A secondary end point was overall survival in the doublet- therapy group as compared with the control group. We report here the results of a prespecified interim analysis.
Overall Survival. Panel A shows the Kaplan–Meier analysis of the probability of survival in the triplet- therapy group as compared with the control group, and Panel B the probability of survival in the doublet-therapy group as compared with the control group. Panel C shows the results of the subgroup analysis of overall survival in the triplet-therapy group as compared with the control group. Eastern Cooperative Oncology Group (ECOG) performance-status scores range from 0 to 5, with higher scores indicating greater disability.
Best Percentage Change in Size of Target Lesions. Shown are the best percentage changes from baseline in the sum of the diameters of the target lesions in each patient in the three groups, as determined by central review. The dashed lines at 20% and −30% indicate progressive disease and partial response, respectively, according to Response Evaluation Criteria in Solid Tumors, version 1.1. The asterisks indicate patients who had a complete response, partial response, or stable disease with respect to target lesions but who had a new lesion, a progressing nontarget lesion, or both.
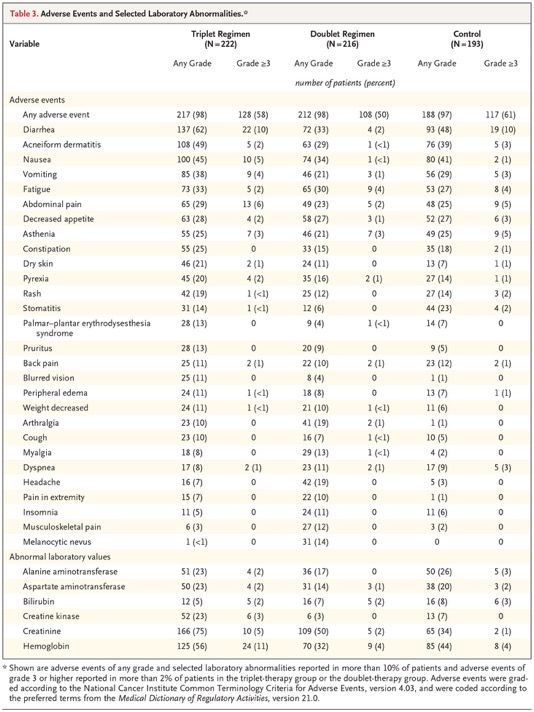
The most common adverse events in the triplet-therapy group were gastrointestinal- related and skin-related events, including diarrhea, nausea, vomiting, and acneiform dermatitis. Low hemoglobin level or anemia was a common laboratory abnormality. Table S3 lists selected adverse events that occurred during the trial, grouped according to clinically similar events that are commonly associated with BRAF and MEK inhibitors. Class-related toxic effects of MEK inhibitors, including serous retinopathy and left ventricular dysfunction, occurred at rates similar to those described previously and were managed with treatment interruptions with or without subsequent dose reduction. Adverse events of grade 3 or higher were observed in 58% of patients in the triplet-therapy group, in 50% in the doublet-therapy group, and in 61% in the control group. Discontinuation of therapy primarily because of an adverse event was seen in 7% of patients in the triplet-therapy group, in 8% in the doublet-therapy group, and in 11% in the control group. Fatal adverse events occurred in 4%, 3%, and 4% of the patients, respectively. Three of the deaths were determined by the investigators to be related to treatment: one death (in the triplet- therapy group) was from colonic perforation, one (in the control group) was from anaphylaxis, and one (in the control group) was from respiratory failure.
Discussion This initial analysis of the BEACON CRC trial shows that the triplet regimen of encorafenib, binimetinib, and cetuximab resulted in longer overall survival and a higher objective response rate than a control regimen of cetuximab plus the investigators’ choice of irinotecan-based chemotherapy in patients with BRAF V600E–mutated metastatic colorectal cancer who had had disease progression after one or two previous regimens. The doublet regimen of encorafenib and cetuximab also resulted in significantly longer overall survival and a higher objective response rate than were seen in the control group. The results in the control group were as expected on the basis of recently reported prospective data in a similar population and several published retrospective analyses. The triplet regimen was designed to be a combination of agents that would provide the most effective inhibition of the MAPK pathway. Preclinical and clinical studies showed that the lack of efficacy of single-agent BRAF or dual BRAF and MEK inhibition in BRAF V600E–mutated colorectal cancer is related to EGFR-mediated adaptive feedback — a finding that led to the development of a combination of BRAF, MEK, and EGFR inhibition through a series of iterative studies. This initial analysis in patients with BRAF V600E–mutated metastatic colorectal cancer who had had disease progression after one or two previous regimens showed that a triplet regimen of encorafenib, binimetinib, and cetuximab or a doublet regimen of encorafenib and cetuximab, as compared with current standard therapy, resulted in a significant and clinically relevant benefit with respect to overall survival and objective response rate. The side-effect profiles of both combination regimens allowed maintenance of high dose intensity for the majority of patients and are consistent with the known profile of each agent. Further follow-up is needed to better define the relative benefits of the triplet and doublet regimens.
Die Spielmeyer-Vogt-Krankheit, auch Batten- Syndrom ist eine sehr seltene angeborene neurodegenerative Erkrankung mit den Hauptmerkmalen ubiquitärer Lipopigmentablagerung in den Lysosomen. Diese zerebellare Form einer Heredoataxie ist die klassische juvenile Form der Neuronalen Ceroid-Lipofuszinosen als CLN3. Die Häufigkeit beträgt in Deutschland bei Geburt etwa 1 zu 143.000, in Schweden etwa 1 zu 45.000. Die Vererbung erfolgt autosomal-rezessiv. Klinische Kriterien sind: Manifestation im 6.–7. Lebensjahr Sehstörung in 70 %, zunehmende Erblindung mit Optikusatrophie, Makuladegeneration und Pigmentablagerungen Verlust erworbener intellektueller und motorischer Funktionen Haltungsschwäche, Gangunsicherheit, Athetose Später Krampfanfälle Überlebensdauer 6–20 Jahre. Im Blutausstrich finden sich Lymphozyten mit großen Vakuolen, eine Diagnosesicherung erfolgt durch Humangenetik.
Als Spleißen bzw. Splicing (englisch splice ‚miteinander verbinden‘, ‚zusammenkleben‘) wird ein wichtiger Schritt der Weiterverarbeitung (Prozessierung) der Ribonukleinsäure (RNA) bezeichnet, der im Zellkern von Eukaryoten stattfindet und bei dem aus der prä-mRNA die reife mRNA entsteht. Die zunächst in der Transkription gebildete prä- mRNA enthält noch Introns und Exons. Durch das Splicing werden die Introns entfernt und die angrenzenden Exons miteinander zur fertigen mRNA verknüpft. Splicing findet zusammen mit der Polyadenylierung (Tailing) des 3'-Endes nach der Transkription statt, ist also ein posttranskriptioneller Vorgang. Im Unterschied dazu ist das Capping des 5'-Endes ein cotranskriptioneller Vorgang. Einige RNAs können Introns ohne die Hilfe eines großen Spliceosoms (siehe unten) entfernen. Die chemische Aktivität dazu besitzen sie selbst, d. h. es handelt sich um Ribozyme, die nur in einigen Fällen (Gruppe II Introns) die Hilfe von Proteinen für eine korrekte Faltung benötigen. Auch bei einigen Krankheitsbildern spielt das Splicing eine große Rolle. Mutationen in introns haben keinen direkten Effekt auf die Sequenz des Proteins, das durch ein Gen codiert wird. In einigen Fällen jedoch betreffen Mutationen Sequenzen, die für das Splicing wichtig sind und führen so zu einem falschen Prozessieren der prä-mRNA. Die so entstehenden RNAs codieren für unfunktionelle oder sogar schädliche Proteine und führen damit zu erblichen Erkrankungen. In den letzten Jahren zeigte sich immer deutlicher, dass Transkription, Prozessierung der RNA (also Splicing, Capping und Tailing), RNA-Export in das Zytoplasma, RNA-Lokalisierung, Translation und RNA- Abbau einander beeinflussen und regulieren.
Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease Genome sequencing is often pivotal in the diagnosis of rare diseases, but many of these conditions lack specific treatments. We describe how molecular diagnosis of a rare, fatal neurodegenerative condition led to the rational design, testing, and manufacture of milasen, a splice-modulating antisense oligonucleotide drug tailored to a particular patient. Proof-of-concept experiments in cell lines from the patient served as the basis for launching an “N-of-1” study of milasen within 1 year after first contact with the patient. There were no serious adverse events, and treatment was associated with objective reduction in seizures (determined by electroencephalography and parental reporting). This study offers a possible template for the rapid development of patient-customized treatments. (Funded by Mila’s Miracle Foundation and others.) A 6-year-old girl presented with the insidious onset of blindness, ataxia, seizures, and developmental regression. The parents’ first concerns dated back to when the girl was 3 years of age, when her right foot began to turn inward. When she was 4 years of age, her family noticed her pulling books close to her face at bedtime. At 5 years of age, she came to medical attention because of modest language and social regression, as well as increased clumsiness and stumbling. In the months before she turned 6 years of age, the progression of symptoms accelerated, and she was hospitalized after the rapid development of loss of vision, frequent falls, dysarthria, and dysphagia. Magnetic resonance imaging (MRI) of the head revealed mild cerebral and cerebellar atrophy, and 24-hour electroencephalography (EEG) revealed several subclinical generalized seizures.
Mutation Identification Patient was heterozygous for c.1102G->C, a known mutation. Whole-genome sequencing was undertaken to search for the missing second mutation, which was hypothesized to be a noncoding mutation or a structural variant missed by standard clinical sequencing. Careful inspection of sequencing data revealed a cluster of chimeric reads deep in MFSD8 intron 6 (Fig. 1B), detected in both the proband and the mother (Fig. S2A). Chimeric reads in this cluster were fused either to unmapped poly-T sequences on one end or to unmapped hexameric repeat sequences (AGAGGG) on the other end, which suggested the presence of a DNA insertion beginning and ending with these motifs. Chimeric breakpoints were offset by 14 bp, suggesting duplication of an endogenous target sequence (Fig. 1B). We deduced that these features were consistent with the insertion of an SVA (SINE–VNTR–Alu) retrotransposon. Indeed, amplification by polymerase chain reaction (PCR) across the breakpoints revealed an approximately 2-kb insertion in the proband and the mother that was confirmed by RepeatMasker analysis to be an SVA retrotransposon. Because SVA insertions have been reported to modulate splicing of nearby genes, we examined splicing patterns in the patient’s family. RNA sequencing (RNA-seq) and reverse-transcriptase–PCR analyses revealed missplicing of exon 6 into a cryptic splice-acceptor site (i6.SA) in MFSD8 intron 6, in a location 119 bp upstream from the SVA insertion site, in blood samples and lymphoblasts from the proband and mother (but not the father or an unaffected sibling). This missplicing precisely segregated with the SVA insertion (Fig. 1C) and Fig. S5) and was predicted to lead to premature translational termination, which supported the pathogenicity of the insertion. Development of an Antisense Oligonucleotide Drug Nusinersen is an FDA-approved antisense oligonucleotide drug for spinal muscular atrophy that changes the splicing pattern of the SMN2 RNA. Reasoning that an antisense oligonucleotide might be similarly used to correct missplicing and restore MFSD8 expression in our patient, we designed antisense oligonucleotides to target the i6.SA cryptic splice-acceptor site and nearby splicing enhancers. When we tested these antisense oligonucleotides in patient fibroblasts, we identified three that boosted normal:mutant splicing ratios by a factor of 2.5 to 3. TY777 was the most efficacious (Figure 2B) and became our lead candidate; we dubbed it “milasen.”
This report shows a path to personalized treatment for patients with orphan diseases. It describes the identification of a novel mutation in a child with neuronal ceroid lipofuscinosis 7 (CLN7, a form of Batten’s disease), a rare and fatal neurodegenerative disease. Identification of the mutation was followed by the development and clinical deployment, within 1 year, of a tailored drug to treat the patient. Study Timeline and Genetic Diagnosis. Panel A shows the timeline from the initial clinical diagnosis to the initiation of treatment. ASO denotes antisense oligonucleotide. Panel B shows an IGV (Integrative Genomics Viewer) image of the patient’s whole-genome sequencing (WGS) read alignments near MFSD8 intron 6. Characters in the mapped sequencing reads indicate mismatched or unaligned (i.e., soft-clipped) bases. Vertical dashed lines indicate chimeric read breakpoints. Panel C shows splicing and translational effects of the SVA (SINE–VNTR–Alu) insertion in MFSD8. The abbreviation “i6” indicates the upstream region of the SVA insertion site in Mutations in the MFSD8 gene intron 6 that is misspliced with exon 6 as a result have been the cause of neuronal of the activation of the i6.SA cryptic splice site by ceroid lipofuscinosis. the SVA insertion.
Antisense Oligonucleotide Drug Development. Panel A shows the location and chemistry of the ASOs that were designed to block the i6.SA splice acceptor site or exonic splicing enhancer (ESE) elements. The ESE elements were predicted with RESCUE-ESE and ESEfinder. 2′-MOE denotes 2′-O-methoxyethyl, and 2′-OMe 2′-O-methyl. Panel B shows the ratio of the normal exon 6–exon 7 (E6–E7) splicing to the abnormal exon 6–intron 6 (E6–i6) splicing (normalized to a no- transfection control), measured in patient fibroblasts that were transfected (for 24 hours at 100 nmol per liter) as indicated. To measure splice isoform-specific levels, multiplex reverse- transcriptase polymerase chain reactions were conducted with isoform-specific primer sets, and then the intensity of the isoform-specific bands was quantified by gel electrophoresis (Fig. S6). “Scrambled” indicates a nontargeting oligonucleotide (TY772). bars indicate 95% confidence intervals of the means. P values were calculated by two-sided t-test. Panel C shows RNA sequencing (RNA-seq) analysis validation of the splice-correcting effect of milasen (TY777). For the calculation of the fraction of normal splicing (exon 6–exon 7), three other splicing events that are mutually exclusive with the normal splicing were considered. Splicing events supported by only one read are not shown. P values were calculated by Fisher’s exact test. Panel D shows intracellular vacuoles, visualized by electron microscopy, in control fibroblasts (MFSD8 wild-type human foreskin fibroblast; BJ cell line) and in patient fibroblasts that are either untreated or transfected with the indicated oligonucleotide. Scoring was performed on a scale of 0 to 5, with 0 representing the lowest and 5 representing the highest level of vacuole accumulation.
N-of-1 Clinical Study. Panel A shows the dosing schedule. (Additional details are provided in Fig. S14A.) Panel B shows the concentration of milasen in cerebrospinal fluid (CSF) before each administration (trough). An additional measurement of the concentration in CSF was obtained at day 174 (without concurrent dose administration). bars indicate the minimum and maximum values of duplicate measurements. Trough levels rose steadily in a dose- proportional fashion until day 40, at which point they dropped to 1.7 ng per milliliter and then resumed their rise with repeated dosing up to a plateau of 18 to 27 ng per milliliter. The dip at day 40 may have been due to a CSF leak, given its coincident timing with a post–lumbar puncture headache after the previous dose. A similar plateauing of CSF trough levels was observed in a previous study of intrathecally delivered nusinersen (9 to 11 ng per milliliter after four repeated doses of 12 mg).8 Panel C shows the trends in seizure frequency and duration as reported in a seizure diary recorded by the parents. Seizures were all of the same type: sudden startle followed by uncontrollable, untriggered laughter that was different from the patient’s natural laugh, at times accompanied by an increase in the nonspecific repetitive hand movements she had at baseline. Panel D shows the trends in seizure activity as detected by electroencephalography (EEG). In a comparison of the means of the initial two recordings and the subsequent three recordings, the daily seizure count, seizure duration, Drug given intrathecally every 14 days and percent cumulative time spent in seizure decreased by 63% (from 31.5 to 11.7 per day), 52% (from 108 seconds to 52 seconds), and 85% (from 3.9% to 0.6%), respectively.
Discussion Over the course of treatment to date, milasen appears to have had an acceptable side-effect profile, with no safety concerns. These findings are consistent with those of previous clinical studies of intrathecal delivery of an FDA-approved antisense oligonucleotide for spinal muscular atrophy and investigational antisense oligonucleotides for amyotrophic lateral sclerosis and Huntington’s disease, which had similarly acceptable side-effect profiles. Treatment has also been accompanied by objective reductions in the frequency and duration of seizures. Milasen itself remains an investigational drug, and it is not suited to the treatment of other patients with Batten’s disease because its design is customized to our patient’s specific mutation. Nonetheless, this experience indicates that antisense oligonucleotides may deserve consideration as a platform for the rapid delivery of individualized treatments. Further exploration of this approach will continue to require careful case-by-case consideration of a number of scientific, clinical, and ethical issues, and expectations need to be tempered by the fact that this approach bears substantial risks. At this time, it should be contemplated only in the context of exceptionally serious or life-threatening circumstances. It should also be recognized that only a minority of patients are likely to have mutations that are amenable to the “mRNA splice- switching” strategy deployed here. More generally, this approach is at present probably scalable to only a limited number of patients, given current limitations on systemic and infrastructural issues (e.g., regulatory burden, manufacturing capacity, cost, and reimbursement). This study illustrates the ability to rationally design, test, and deploy a novel therapeutic agent for a patient with a rare disease on the basis of an understanding of her specific pathogenic mutation. It is an example of individualized genomic medicine.
A 77-year-old right-handed woman with limited cutaneous systemic sclerosis presented to the rheumatology clinic with a 2-year history of slowly progressive, painful swelling of her fingertips. On physical examination, the patient had telangiectases on her face and thorax and thickening of the skin on her right hand, both forearms, and face, as well as soft-tissue swelling of the tips of the fingers of both hands (Panels A and B). Findings on capillaroscopy were notable for disorganization of capillaries, avascular areas, and the presence of enlarged capillaries. A radiograph of the hands showed calcifications near the distal phalanx of the first, second, and third fingers of the right hand and the first finger of the left hand (Panel C), which were consistent with calcinosis, a manifestation of limited cutaneous systemic sclerosis. These calcium deposits may grow, ulcerate, and become infected. Despite trials of multiple medications, the patient had progressive pain and numbness in her fingers, with increasing difficulty handling objects, and was referred for surgical evaluation regarding excision of the lesions.
A 60-year-old woman who was a current smoker presented to the emergency department with acute chest pain. The troponin I level was 51 ng per liter on a high-sensitivity assay (reference value,
Der Von-Willebrand-Faktor (VWF) oder von-Willebrand-Faktor (vWF) ist ein Protein, das als Trägerprotein des Blutgerinnungsfaktors VIII eine wichtige Rolle bei der Blutstillung spielt und nach Erik Adolf von Willebrand (1870– 1949), einem finnischen Internisten, benannt wurde. Der Von-Willebrand-Faktor wird sowohl von Megakaryozyten als auch von den Endothelzellen gebildet, die die Innenwand eines Blutgefäßes (die Intima) bilden. In den Blutplättchen (Thrombozyten) als Abschnürungen der Megakaryozyten ist er in den α-Granula gespeichert, das Gefäßendothel gibt ihn zur Speicherung an die subendotheliale Matrix ab. Kommt es zu einem Riss der Innenwand des Endothels, werden die darunter liegenden Proteine der Gefäßwand freigelegt u. a. Kollagene. An diese kann der Von-Willebrand-Faktor binden. Bestimmte zelluläre Elemente des Blutes, die Blutplättchen (Thrombozyten), verfügen auf ihrer Oberfläche über eine Andockstelle, an die der Von-Willebrand-Faktor binden kann. Diese wird als Von-Willebrand-Rezeptor oder Glykoprotein Ib/V/IX bezeichnet. Der Von-Willebrand-Faktor schafft also eine Brücke zwischen den Blutplättchen und der verletzten Gefäßwand. An bereits adhärenten Thrombozyten kann der vWF mittels Glykoprotein IIb bzw. IIIa binden. Der Von- Willebrand-Faktor hat somit eine direkte Wirkung bei der zellulären Blutstillung. Bei der plasmatischen Blutgerinnung ist er zwar nicht direkt beteiligt, da er zur Bildung des Fibrins nicht erforderlich ist. Durch seine Funktion als Träger- und Schutzprotein für den Gerinnungsfaktor VIII führt ein Mangel an Von-Willebrand-Faktor oder ein Defekt seiner Proteinstruktur durch diese unmittelbare Wechselwirkung aber dennoch zur Beeinträchtigung der plasmatischen Hämostase (s. Willebrand- Jürgens-Syndrom und von-Willebrand-Krankheit).
N Engl J Med 2019 earlier
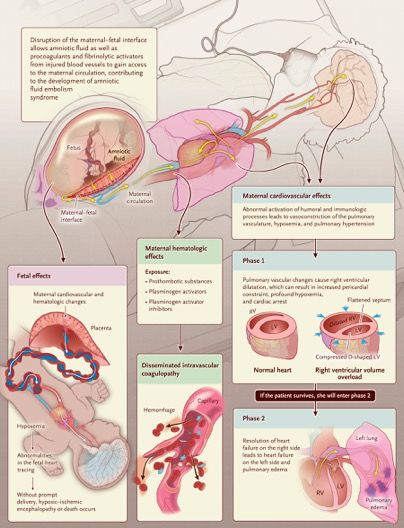
Pathophysiology of Severe ADAMTS13 Deficiency. In an arteriole of a healthy person (Panel A), von Willebrand factor is synthesized by endothelial cells and stored in Weibel–Palade bodies as long strings of repeating subunits. After stimulation of endothelial cells, these ultralarge von Willebrand factor multimers are secreted and some of these molecules remain anchored to the endothelial-cell surface; others are released into the circulation. Flowing blood unfolds ultralarge von Willebrand factor multimers, exposing the platelet binding and the ADAMTS13 cleavage sites. After ADAMTS13 cleavage, von Willebrand factor molecules again adopt a coiled, condensed shape without exposed platelet binding or ADAMTS13 cleavage sites. In the absence of ADAMTS13 (Panel B), many platelets attach to the exposed platelet-binding sites on the ultralarge von Willebrand factor multimer strings. Some of these multimers remain attached to the endothelial surface, coating the vessel wall. Other ultralarge von Willebrand factor strings detach from the endothelial-cell surface and enter the circulation, where they become coated with adherent platelets. As these platelet-loaded ultralarge von Willebrand factor multimers approach small vessels and bifurcations with sharp turns, blood flow accelerates, increasing shear stress. Increased shear stress causes further unfolding of the ultralarge von Willebrand factor molecules, exposing more platelet-binding sites. In conditions involving increased von Willebrand factor synthesis and release into plasma (e.g., during infection, inflammation, and pregnancy), the circulating ultralarge von Willebrand factor molecules with their many attached platelets form webs at the sites of increased shear stress, obstructing blood flow and causing thrombosis
Hereditary Thrombotic Thrombocytopenic Purpura Hereditary thrombotic thrombocytopenic purpura (TTP), also known as Upshaw–Schulman syndrome (Online Mendelian Inheritance in Man number, 274150), is a rare autosomal recessive disorder caused by ADAMTS13 mutations that result in the absence or severe deficiency of the plasma metalloprotease ADAMTS13. ADAMTS13 is required for cleavage of newly synthesized von Willebrand factor multimers. Decreased ADAMTS13 activity is associated with an increased size of von Willebrand factor multimers and an increased risk of microvascular thrombosis. Patients with hereditary TTP may appear to be healthy, but their increased risk of critical thrombosis is always present. This review describes the history, pathogenesis, prevalence, clinical features, and current management of hereditary TTP, as well as potential future treatments. The only known substrate of ADAMTS13 is von Willebrand factor, which is exclusively synthesized by endothelial cells and megakaryocytes.
ADAMTS13 Mutations in Hereditary TTP More than 200 ADAMTS13 mutations, spread over all ADAMTS13 protein domains, have been identified in patients with hereditary TTP. In addition, there are at least 10 missense ADAMTS13 single-nucleotide polymorphisms, some of which are in strong linkage disequilibrium with specific ADAMTS13 mutations and influence their molecular effects. Clinical Features Some patients with hereditary TTP may have symptoms that manifest at birth, whereas others remain asymptomatic for decades. When symptoms occur, the clinical features may be indistinguishable from an acute episode of acquired TTP or they may differ substantially. Typical Triggers of Disease Manifestations and Heterogeneous Clinical Presentations in Hereditary TTP. Shown are four patients with hereditary TTP who present at times of risk (i.e., the neonatal period, after heavy alcohol intake, and during pregnancy) or with manifestations that occur spontaneously. All the patients have common clinical features that are distinct from those that are characteristic of patients with acquired TTP. Some patient data are courtesy of Eric Avery, M.D., Lincoln, Nebraska. CT denotes computed tomography, and MRI magnetic resonance imaging.
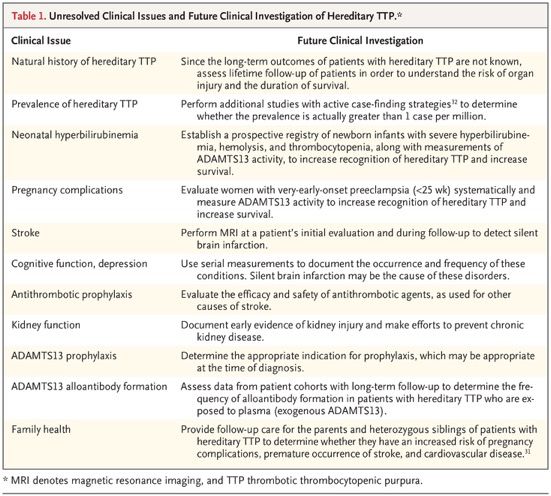
Distinguishing Hereditary TTP from Acquired TTP The patient’s age may help to distinguish between hereditary TTP and acquired TTP. Acquired TTP is much less common in young children than in adults, whereas hereditary TTP is commonly diagnosed in children. In the Japanese cohort, the disease first manifested in 33 of the 43 patients (77%) before 10 years of age. The presence of a functional ADAMTS13 inhibitor or an increased anti–ADAMTS13 IgG antibody titer argues against the diagnosis of hereditary TTP. An exception is the occurrence of severe hemolysis with a high plasma hemoglobin concentration (≥2 g per deciliter), which can cause false positive results in functional ADAMTS13 inhibitor assays.58 Although the absence of a functional ADAMTS13 inhibitor is characteristic of hereditary TTP, it does not rule out the diagnosis of acquired TTP. Among patients with acquired TTP in the Oklahoma Thrombotic Thrombocytopenic Purpura–Hemolytic Uremic Syndrome Registry, a functional ADAMTS13 inhibitor was not initially identified in 15 of 86 patients (17%). Long-Term Outcomes Data on long-term outcomes in patients with hereditary TTP are lacking. In the Japanese cohort of 43 patients, only 13 patients (30%) were followed after 35 years of age, and only 6 (14%) were followed after 45 years of age. Three of the 43 patients (7%) died at the ages of 38 to 79 years — 2 from chronic kidney disease and 1 from stroke. In the Norwegian cohort of 21 patients (including 3 siblings who were probably affected), 5 (24%) died. Two of these patients died before 1 year of age, and the other 3 died at 40 to 60 years of age. The causes of death were not reported. In the cohort in the United Kingdom, 4 of the 73 patients (5%) died from stroke; the patients’ ages were not reported. Five of the 120 patients (4%) in the International Hereditary Thrombotic Thrombocytopenic Purpura Registry died; their ages and causes of death were not reported. Current Management Plasma infusion is usually sufficient to treat acute episodes in patients with hereditary TTP, although for severe manifestations (e.g., in pregnancy) therapeutic plasma exchange may be appropriate. For patients who have recurrent symptoms, regular lifetime prophylactic plasma infusions are appropriate, although the decision to begin prophylaxis is difficult, especially in a child. The half-life of ADAMTS13 activity in patients receiving regular prophylactic plasma infusions is 2.5 to 3.5 days. The development of inhibitory ADAMTS13 alloantibodies in patients receiving plasma therapy has been reported twice. Future Management of Hereditary TTP The availability of rhADAMTS13 may revolutionize the management of hereditary TTP.
A 35-Year-Old Woman with Cardiopulmonary Arrest during Cesarean Section At 6 weeks 4 days of gestation, the patient had been seen at a community health center because of a positive pregnancy test. She was gravida 4, 1-0-2-1. Her first pregnancy, 5 years earlier, had resulted in the need for a cesarean section because the fetus was in the breech presentation; the baby was delivered at full term without complications. The second and third pregnancies had been electively terminated. The patient reported that her husband had human immunodeficiency virus type 1 (HIV-1) infection and that he had been taking antiretroviral therapy; he had had undetectable plasma levels of HIV-1 RNA during the 5 months before this visit. Blood testing in the patient for HIV-1 and HIV-2 antibodies and HIV-1 p24 antigen with the use of a fourth-generation combination assay was negative. Preexposure prophylaxis with a fixed-dose combination of emtricitabine and tenofovir disoproxil fumarate was prescribed. Twenty-five days before presentation, at 33 weeks of gestation, vaginal bleeding developed, and the patient was brought by ambulance to the obstetrical unit of this hospital. She reported that on awakening that morning, she had passed a large volume of bright red, liquid blood and three large clots, each measuring approximately 1.5 cm in diameter, through the vagina. She felt occasional contractions; fetal movement was normal. She reported no recent trauma, fall, or sexual intercourse. Ultrasonographic examination was performed; the placenta was positioned anteriorly along the midline of the uterus, completely overlying the cervical os, and there was no evidence of placenta accreta.
The patient underwent cesarean section under combined spinal–epidural anesthesia. Cefazolin was administered intravenously before incision of the skin. During the surgery, a low transverse uterine incision was made through the anterior placenta. The membranes were ruptured bluntly, yielding clear amniotic fluid. The uterine incision was extended, and the infant was delivered 37 minutes after injection of the spinal anesthetic agent, which was administered during a combined spinal–epidural procedure; the opaque surgical drape was lowered, allowing the patient to see the infant through the clear drape. The 1-minute and 5-minute Apgar scores were 8 and 10, respectively. The placenta was removed with gentle traction. Three minutes after delivery of the infant, while clot and debris were being cleared from the uterine cavity, the exteriorized uterus became blanched with minimal bleeding. Concurrently, the patient was noted to be unresponsive, with sporadic respirations; carotid and aortic pulses were present. Manual ventilation was begun. Two minutes later, the patient became pulseless, with normal sinus rhythm seen on the patient monitor of the anesthesia machine. Cardiopulmonary resuscitation was begun, the trachea was intubated, and mechanical ventilation was initiated.
Pulmonary Embolism Did this patient have a pulmonary embolism that led to cardiovascular collapse? In support of this diagnosis was the acute nature of the event and the fact that the patient had several risk factors for pulmonary embolism, including hypercoagulability, pregnancy, advanced maternal age, and obesity. Hemorrhage The patient had multiple risk factors for hemorrhage, and this was the adverse outcome for which we were most prepared. Given that she had had a previous cesarean section and that she had a complete anterior placenta previa in this pregnancy, we had concerns about a potential placenta accreta. Venous Air Embolism Venous air embolism has been reported to occur at the time of cesarean section. This diagnosis should be considered any time the operative site is positioned above the patient’s heart, which occurred in this patient when the uterus was exteriorized. However, as is the case with pulmonary embolism, these events are often preceded by wheezing, dyspnea, chest pain, or gasping. Anaphylaxis Anaphylaxis was also a consideration in this case, given that many medications were administered before and during the procedure. However, the patient did not have any known drug allergies, and the timing for an anaphylactic event did not fit well, since cardiovascular collapse occurred more than 30 minutes after these treatment exposures. High Cephalad Spread of Neuraxial Anesthetic Agent High cephalad spread of a neuraxial anesthetic agent can cause sudden loss of consciousness and cardiac arrest, but in this case, the event occurred more than 30 minutes after injection of the spinal anesthetic agent, which was administered during a combined spinal–epidural procedure. Peripartum Cardiomyopathy and Myocardial Infarction The patient had risk factors for peripartum cardiomyopathy, including being in the late stage of pregnancy, being of advanced maternal age, and being of African descent. Eclampsia Eclampsia should be considered in any pregnant woman who has an acute change in mental status. However, this patient was normotensive and did not report headaches, vision changes, or pain in the right upper quadrant. Also, she did not show any signs of seizure activity.
Sie können auch lesen