Autosomal dominante nicht syndromale Hörstörungen
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Genetik der HNO Krankheiten
Autosomal dominante nicht syndromale Hörstörungen
Christian Kubisch
Institut für Humangenetik
Universitätsklinikum Bonn
Zusammenfassung Summary Einleitung
Im Gegensatz zu den autosomal re Autosomal dominant hearing Bei den genetisch bedingten Hör
zessiven nichtsyndromalen Hör impairment is not as frequent as störungen handelt es sich um eine
störungen sind die autosomal domi autosomal recessive hearing loss and Gruppe von z.T. sehr unterschiedli
nanten Formen seltener und meist is most often characterized by a later chen Erkrankungen, die z.B. nach
durch einen späteren Krankheitsbe age of onset and a progressive dem Beginn des Auftretens, der mög
ginn und einen fortschreitenden Cha course of the disease. So far, more lichen Progredienz, dem Schweregrad
rakter gekennzeichnet. Bisher sind than 35 independent loci have been des Hörverlusts, dem Vorliegen weite
für die autosomal dominanten For mapped for autosomal dominant rer Symptome im Sinne eines defi
men mehr als 35 unabhängige Loci forms and 15 genes have been nierten Syndroms oder aber dem Ver
kartiert und 15 Gene identifiziert wor identified, yet, in most cases erbungsmodus klassifiziert werden
den, wobei Mutationen meist in nur mutations have been found in just a können. Dieser Artikel beschäftigt sich
wenigen Familien gefunden werden few families. Some of the identified mit den autosomal dominanten nicht
konnten. Einige dieser Gene sind genes are also responsible for syndromalen Hörstörungen (ADNSHL)
auch für autosomal rezessive Formen autosomal recessive forms or und versucht einen allgemeinen Über
bzw. syndromale Hörstörungen ver syndromic hearing loss. The function blick über diese heterogene Gruppe
antwortlich. Die Funktion der betrof of the responsible genes is von Erkrankungen zu geben.
fenen Gene ist außerordentlich viel extraordinary diverse and reflects the
fältig und reflektiert die Komplexität complexity of the structure and Häufigkeit
des Aufbaus und der Funktion des function of the inner ear. In particular, Bei den prälingualen Formen der erb
Innenohrs. Insbesondere sind dabei genes encoding for proteins of the lichen Hörstörungen werden ca. 80 –
Gene identifiziert worden, die für cytoskeleton and extracellular matrix 85% autosomal rezessiv und ca. 15%
Proteine des Zytoskeletts und der as well as transcription factors and autosomal dominant vererbt. Bei den
Extrazellulärmatrix sowie für Trans ion transport proteins have been später manifesten Formen liegen kei
kriptionsfaktoren und Ionentransport found. The results of the genetic ne genaueren Einschätzungen hin
proteine kodieren. Durch die geneti studies have improved our sichtlich der Häufigkeit der verschie
schen Befunde konnte das Verständ understanding about the denen Vererbungsmodi vor, dennoch
nis über die Entwicklung des Ohres development of the ear and the ist anzunehmen, dass hier der Anteil
und die Physiologie des Hörens er physiology of hearing. der autosomal dominanten Formen
weitert werden. höher ist. Insgesamt kann vereinfa
chend gesagt werden, dass die auto
Schlüsselwörter Keywords somal rezessiven Formen meist
Schwerhörigkeit, autosomal domi Hearing loss, autosomal dominant schwerer in ihrer klinischen Sympto
nanter Erbgang mode of inheritance matik sind und einen Großteil der kon
genitalen Fälle der Taubheit ausma
chen. Die Häufigkeit einer kongenita
len Taubheit wird mit ca. 1:1 000 an
gegeben (Marazita, 1993). Demge
genüber sind die meisten autosomal
dominanten Formen durch eine spä
tere Manifestation (oft im frühen Er
wachsenenalter) und einen progre
dienten Charakter gekennzeichnet,
30 medgen 14 (2002)Genetik der HNO Krankheiten
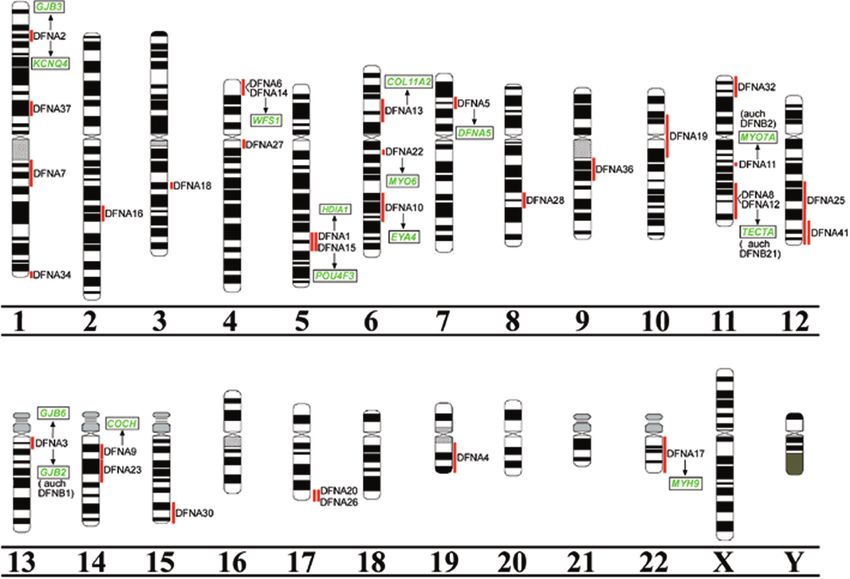
Abb 1
Publizierte Genloci für die autosomal domi
nanten nicht syndromalen Hörstörungen,
dargestellt anhand von Ideogrammen
menschlicher Chromosomen.
Die roten Balken neben den Ideogrammen re
präsentieren die ungefähre zytogenetische Lo
kalisation der DFNALoci. In grüner Schrift sind
die identifizierten Gene dargestellt, auf die im
Text detailliert eingegangen wird. In den Fällen,
in denen ein ADNSHLGen auch in rezessiven
Formen gefunden worden ist, ist dies in Klam
mern unter oder über dem Gen angegeben, wo
bei der entsprechende DFNBLokus benannt
worden ist. Xchromosomale Loci sind nicht ge
zeigt.
wenngleich auch hier schwere, nicht Kopplungsanalysen und Loci für 2002). Die exakte Anzahl an Loci kann
progrediente und kongenitale Formen die ADNSHL nicht genannt werden, da
vorkommen. Dies ist z.B. bei den For Schon vor der ersten erfolgreichen
men DFNA3 und DFNA8/12 bekannt, Identifizierung eines „Taubheitsgens“ (1) in einigen Fällen Loci als unabhän
wobei DFNA die Bezeichnung für ei war bekannt, dass sowohl autosomal gig beschrieben wurden, sich aber
nen autosomal dominanten Lokus ei dominante als auch autosomal rezes nach der Genidentifizierung her
ner nichtsyndromalen Hörstörung ist sive nichtsyndromale Hörstörungen ausstellte, dass dasselbe Gen zu
und die nachfolgende Zahl einen spe insofern außergewöhnlich sind, als für grunde lag (z.B. DFNA8/12 und
zifischen Lokus beschreibt. Die Lo diese Erkrankungen eine extreme He DFNA 6/14),
kusnummern werden chronologisch terogenie vorausgesagt worden war.
nach der Erstbeschreibung vergeben. Konkret wurde geschätzt, dass Verän (2) auch weitere Loci, für die das Gen
derungen in bis zu 100 verschiedenen noch nicht bekannt ist, überlappen,
Betrachtet man weiterhin die Präva Genen für eine monogene, nichtsyn und somit z. Zt. nicht klar ist, ob
lenz von Hörstörungen im fortge dromale Hörstörung verantwortlich zwei unterschiedliche Gene betrof
schrittenen Erwachsenenalter, so zei sein können (Morton, 1991). fen sein könnten,
gen zwischen dem 40. und 50. Le
bensjahr bzw. zwischen dem 60. und Eine Genidentifizierung bei der ADNS (3) in einigen Fällen an einem Lokus
70. Lebensjahr 0,3% bzw. 2,3% der HL setzt voraus, dass Familien identi mehr als ein ursächliches Gen in
Menschen eine hochgradige Schwer fiziert und analysiert werden, in denen unterschiedlichen Familien gefun
hörigkeit (d.h. ein Hörverlust größer ein Hörverlust autosomal dominant den worden ist (z.B. DFNA2) und
65 dB), wobei die Hörleistung im we vererbt wird und die groß genug sind,
niger stark betroffenen Ohr zugrunde um eine genomweite Kopplungsana (4) einige reservierte Loci bisher nicht
gelegt wird. Schließlich liegt zwischen lyse mit ausreichender statistischer publiziert worden sind und somit
dem 70. und 80. Lebensjahr bei mehr Aussagekraft durchführen zu können. eine mögliche Parallelbeschrei
als 60% der Bevölkerung eine Hör Nach der Identifizierung des genomi bung oder Überlappung mit be
minderung von >25 dB vor (Davis, schen Lokus werden dann Gene aus kannten Loci nicht letztlich auszu
1989). Die Altersschwerhörigkeit ist im dieser Region auf das Vorliegen von schließen ist.
überwiegenden Teil der Fälle nicht Mutationen untersucht, um somit das
monogen, sondern multifaktoriell be ursächliche Gen für die ADNSHL zu Die bisher publizierten Loci der AD
dingt. Neben Umweltfaktoren wie z.B. finden. Dieses Vorgehen der sog. „Po NSHL sind in Abbildung 1 schema
Lärm und ototoxischen Medikamenten sitionellen Klonierung“, bei dem a pri tisch gezeigt, wobei bereits identifi
spielen jedoch auch hier genetische ori nichts über die Funktion des ver zierte Gene ebenfalls dargestellt sind.
Faktoren eine bedeutende Rolle. Hin antwortlichen Gens bekannt ist, war in Insgesamt handelt es sich bei den
sichtlich der Aufklärung dieser bis den letzten Jahren bei der Aufklärung ADNSHL meist um später manifeste
jetzt nicht bekannten genetischen Prä von genetischen Loci bzw. Genen der Erkrankungen (Ausnahme DFNA3 und
dispositionsfaktoren spekuliert man, ADNSHL sehr erfolgreich. Bisher sind DFNA8/12 mit kongenitalem Beginn),
dass insbesondere die Gene der AD mehr als 35 unabhängige Loci für die wobei die hauptsächlich betroffenen
NSHL und der mitochondrialen Hör ADNSHL beschrieben (s. a. die stän Frequenzen in den Familien unter
störungen gute Kandidatengene für dig aktualisierten Tabellen auf der schiedlich sind, worauf hier jedoch
die Presbyakusis sind. „Hereditary Hearing Loss Homepage“ nicht eingegangen werden kann.
unter http://dnalabwww.uia.ac.be/
dna lab/hhh) (Van Camp and Smith,
medgen 14 (2002) 31Genetik der HNO Krankheiten Tab 1 Tabellarischer Überblick über die bekannten ADNSHL Gene und ihre Funktion,
die vermutete Häufigkeit von Mutationen dieser Gene und mögliche allelische Erkrankungen
Loci und bekannte Gene für die ADNSHL Häufigkeit Besonderheiten Funktion
DFNA1: HDIA1 selten Aktinorganisation
DFNA2: Connexin 31 selten Allelie mit ARNSHL und Kaliumrecycling?
Erythrokeratodermia variabilis
(ohne Hörstörung)
DFNA2: KCNQ4 wahrscheinlich Kaliumrecycling? Modulation der
etwas häufiger Erregbarkeit der Haarzellen?
DFNA3: Connexin 26 selten Allelie mit ARNSHL (DFNB1) Kaliumrecycling
DFNA3: Connexin 30 selten Allelie mit ARNSHL und der hidro Kaliumrecycling?
tischen ektodermalen Dysplasie
(ohne Hörstörung)
DFNA5: DFNA5 selten unbekannt
DFNA6/14/38: Wolframin wahrscheinlich häufig Allelie mit WolframSyndrom unbekannt
bei Tieftonschwer (autosomal rezessiv)
hörigkeit
DFNA8/12: TECTA selten kongenital und nichtprogrediente Aufbau der Tektorialmembran
Hörstörung
DFNA9: COCH nicht bekannt Assoziation mit M. Menière ExtrazellulärmatrixProtein?
Innenohrentwicklung ?
DFNA10: EYA4 selten
DFNA11: MYO7A selten Allelie mit ARNSHL (DFNB2) und Zytoskelett
UsherSyndrom (USH1B)
DFNA13: COL11A2 selten Allelie mit SticklerSyndrom Aufbau der Tektorialmembran
DFNA15: POU4F3 selten Haarzellentwicklung ?
DFNA17: MYH9 selten Allelie mit MayHegglinAnomalie, Zytoskelett
FechtnerSyndrom, Sebastian
Syndrom, EpsteinSyndrom und
AlportSyndrom mit Makrothrom
bozytopenie
DFNA22: MYO6 selten Zytoskelett
Überschneidung mit autosomal Bisher konnten 15 Gene für die AD laubte, einen systematischen, geno
rezessiven und syndromalen NSHL identifiziert werden, die hier mischen Sequenzierungsansatz zu
Hörstörungen kurz vorgestellt werden. Insgesamt verfolgen. In diesem Fall konnte 1997
Neben der ausgeprägten Heterogenie kann aufgrund der Platzlimitierung je ein kochleär exprimiertes Gen identi
zeigte sich schon bald nach der Be doch nur ein allgemeiner Überblick fiziert werden, das eine signifikante
schreibung der ersten Loci und Gene über die komplexen genetischen Be Homologie zu bekannten Genen aus
ein weiterer, zusätzlich komplizieren funde gegeben werden, auch hin anderen Spezies aufwies, nämlich
der Aspekt hinsichtlich der geneti sichtlich der Quellen der beschriebe dem diaphanousGen aus der Frucht
schen und klinischen Klassifikation nen Ergebnisse soll nochmals auf die fliege Drosophila melanogaster und
von Hörstörungen. So fand sich, dass „Hereditary Hearing Loss Homepage“ dem Gen p140mDia aus der Maus.
Gene, die bei der ADNSHL mutiert verwiesen werden. Das humane Ortholog wurde als diap
sind, auch ursächliche Veränderungen hanous 1 (HDIA1) bezeichnet und
in autosomal rezessiven Formen Bisher bekannte Gene tatsächlich konnte in der untersuchten
und/oder syndromalen Formen der für die ADNSHL Familie eine Mutation entdeckt wer
Hörstörungen (bzw. sogar Syndromen den, die mit der Krankheit kosegre
ohne Hörstörungen) aufwiesen, d.h. DFNA1: gierte und zu einer Proteintrunkation
es liegt zumindest in einem Teil der HDIA1 und Aktinpolymerisation führt (Lynch, 1997). Dieses Protein
Fälle eine Allelie von klinisch bzw. ge Der erste autosomal dominante Taub gehört zur sog. FH (Formin Homolo
netisch unterschiedlichen Erkrankun heitslokus, DFNA1, konnte 1992 in ei gie) Protein Familie, deren Mitglieder
gen vor. Auf diese Überschneidungen ner großen Familie aus Costa Rica auf eine zentrale Rolle in der Regulation
und mögliche pathophysiologische Er Chromosom 5q31 kartiert werden. In der Aktinorganisation einnehmen. Man
klärungsansätze hierfür wird im fol nachfolgenden Arbeiten gelang es, die nimmt heute an, dass das humane
genden bei der Beschreibung der be chromosomale Region, in der das ver HDIA1 wahrscheinlich an der Aktinor
reits identifizierten Gene kurz einge antwortliche Gen liegen musste, auf ganisation innerhalb der Stereozilien
gangen. ca. 800 kb einzugrenzen. Somit war oder in der Kutikularplatte, die beson
eine Größe erreicht worden, die es er ders reich an Aktinpolymeren ist, be
32 medgen 14 (2002)Genetik der HNO Krankheiten
teiligt ist. Störungen dieser Funktion Das andere DFNA2Gen ist der span scheinlich relativ begrenzt. Dominan
führen wahrscheinlich zum Verlust der nungsabhängige Kaliumkanal KCNQ4, te Mutationen zeigen in der elektro
Haarzellstruktur und somit der Fähig der ein Homolog des KCNQ1Gens physiologischen Untersuchung einen
keit zur mechanoelektrischen Signal ist, welches u. a. bei der syndromalen dominantnegativen Effekt im Gegen
transduktion. Hörstörung des JervellLangeNiel satz zu der Haploinsuffizienz rezessi
senSyndroms mutiert ist (Taubheit ver Mutationen, was die unterschied
DFNA2: und Herzrhythmusstörung im Sinne ei lichen Erbgänge in Abhängigkeit von
KCNQ4, Connexin 31 und mehr? nes „Long QTSyndroms“). Nach der der zugrunde liegenden Mutation er
Bei der Kartierung des DFNA2Lokus Klonierung und chromosomalen Kar klären kann.
auf Chromosom 1p34 wurden 1994 tierung von KCNQ4 auf dem kurzen
positive Kopplungsergebnisse für Arm von Chromosom 1 konnte durch Da einige zum DFNA3Lokus kartierte
mehrere Familien gefunden, die so in eine in situHybridisierung gezeigt Familien keine Mutation im GJB2Gen
terpretiert wurden, dass alle Familien werden, dass KCNQ4 im Innenohr zeigten und da das GJB6Gen sich
Mutationen im selben Gen haben. spezifisch in den äußeren Haarzellen ebenfalls auf Chromosom 13q12 be
Aufgrund der Heterogenie der nicht lokalisiert ist. Die chromosomalen und findet, wurde auch dieses Connexin
syndromalen Hörstörungen war dies histologischen Befunde machten den gen für die ADNSHL untersucht und
jedoch eine unzulässige Schluss Kaliumkanal zu einem attraktiven Kan tatsächlich fand sich auch im GJB6
folgerung, wie sich 1999 zeigte. So didaten für DFNA2. Tatsächlich konn Gen in einer Familie eine Mutation
wurden in verschiedenen Familien te in einer Familie mit autosomal do (Grifa, 1999). Auch diese Mutation
Mutationen in zwei verschiedenen Ge minanter Hörstörung eine Mutation in zeigte in der elektrophysiologischen
nen innerhalb des DFNA2Lokus ge der hochkonservierten Porenregion Untersuchung einen dominantnegati
funden. In einer Familie, die ebenfalls des Kanals identifiziert werden (Ku ven Effekt, der die Pathogenität be
auf diesen Lokus kartiert, konnte kei bisch, 1999). Elektrophysiologische weist. In Analogie zum GJB3 fanden
ne Mutation in den beiden Genen Analysen zeigten einen dominant ne sich auch für dieses Connexingen
identifiziert werden, so dass davon gativen Effekt der Mutante auf den weitere Mutationen in Familien mit
ausgegangen wird, dass noch minde Wildtypkanal, so dass die Funktion ARNSHL (del Castillo, 2002) und bei
stens ein weiteres „Taubheitsgen“ auf des Kaliumkanals, der wahrscheinlich einer dermatologischen Erkrankung,
1p34 lokalisiert ist (Van Hauwe, 1999). als Tetramer aufgebaut ist, um fast der hidrotischen ektodermalen Dys
90% reduziert wurde. Nachfolgend plasie (Lamartine, 2000).
Das eine bekannte DFNA2Gen wurde konnten auch unabhängige Arbeits
in einem Kandidatengenansatz auf gruppen mehrere Familien identifizie DFNA5 – Funktion unbekannt
grund der zentralen Bedeutung von ren, in denen KCNQ4Mutationen vor Ein Großteil der bekannten „Taub
Gap Junctions im Innenohr gefunden, lagen. Die Funktion dieses Kanals ist heitsgene“ ist durch eine positionelle
wobei es sich um das sog. Connexin z. Zt. unklar. Aufgrund seiner Expres Klonierungsstrategie gefunden wor
31 (GJB3 oder auch CX31) handelte. sion in den sensorischen äußeren den. Wenngleich die Funktion der mei
Tatsächlich gelang es, Mutationen im Haarzellen kann spekuliert werden, sten Gene durch funktionelle Studien
GJB3, das im Innenohr exprimiert ist, dass er an der basolateralen Kalium (z.B. Connexine und Kaliumkanäle)
in zwei Familien aus China mit auto leitfähigkeit der Haarzellen beteiligt oder Sequenzvergleiche mit bereits
somal dominant vererbter Hörstörung sein könnte, die sowohl für die Modu bekannten Genen (z.B. Myosine,
nachzuweisen (Xia, 1998). Einige der lation der elektrischen Erregbarkeit als HDIA1 und Tektorine) mit großer
Trägerinnen der Mutationen sind nur auch für die Entfernung des apikal Wahrscheinlichkeit vorhergesagt wer
subklinisch oder gar nicht von einem aufgenommenen Kaliums wichtig ist. den kann, ist ein immanentes Problem
Hörverlust betroffen, so dass von ei der positionellen Klonierung, dass un
ner inkompletten Penetranz oder (ge DFNA3: ter Umständen Gene gefunden wer
schlechtsspezifisch) modifizierenden Ein Lokus und zwei Connexin Gene den, über deren Funktion nichts be
Faktoren ausgegangen werden kann. Auch am DFNA3Lokus konnten zwei kannt ist. Bei diesen Genen kann le
Mutationen im GJB3 finden sich auch „Taubheitsgene“ gefunden werden, diglich aus Mutationsanalysen in
in Familien mit autosomal rezessiven wobei es sich um zwei weitere Con „Taubheitsfamilien“ und der Lokalisa
nichtsyndromalen Hörstörungen (Liu, nexinGene handelt, das Connexin 26 tion des Gens im Innenohr geschlos
2000). Eine weitere Studie konnte zei (GJB2 oder CX26) und das Connexin sen werden, dass es für eine familiäre
gen, dass andere Mutationen im GJB3 30 (GJB6 oder CX30). Eine detaillierte Hörstörung verantwortlich ist. In die
zu einer autosomal dominanten Haut Beschreibung des GJB2Gens und se Kategorie fällt z.B. das DFNA5
erkrankung, der Erythrokeratodermia seiner großen Bedeutung für die auto Gen, das nach seinem Lokus benannt
variabilis, führen (Richard, 1998). Wei somal rezessiven nichtsyndromalen worden ist. Für diesen ADNSHLLo
tere Studien werden zeigen, ob und Hörstörungen findet sich in dem ent kus auf dem kurzen Arm von Chromo
inwieweit Patienten mit GJB3 beding sprechenden Beitrag dieser Ausgabe. som 7 konnte 1998 ein Gen identifi
ter Hörstörung bzw. Hauterkrankung Die Bedeutung des GJB2 für die AD ziert werden, dessen Mutation perfekt
auch subklinische Zeichen der ent NSHL (Denoyelle, 1998) ist nach an mit der Krankheit in einer Familie ko
sprechend anderen Erkrankung auf fänglichen Zweifeln unbestritten, je segregierte (Van Laer, 1998). Da keine
weisen. doch hinsichtlich der Häufigkeit wahr signifikante Homologie zu bekannten
medgen 14 (2002) 33Genetik der HNO Krankheiten
Genen gefunden werden konnte, ist bindungen, d.h. es wird vermutlich ein le mit M. Menière verantwortlich sind,
die Funktion des Gens, das im Inne dominant negativer Effekt auftreten, ist nicht bekannt.
nohr exprimiert ist, unklar. der auch den dominanten Erbgang er
klären kann. Auffälligerweise und un DFNA10 – EYA4, ein Homolog des
DFNA6/14/38 – ADNSHL und das typisch früh für eine dominante Taub „Eyes absent“ Gens
Wolfram Syndrom heitsform ist der prälinguale Beginn Die EYAGene gehören zu einer Fami
Drei Loci, DFNA6, DFNA14 und der Erkrankung in beiden Familien. Als lie von transkriptionellen Aktivatoren
DFNA38, wurden auf den kurzen Arm mögliche Erklärung hierfür ist jedoch in Vertebraten und das EYA1Gen ist
von Chromosom 4 kartiert, wobei zu die Tatsache geeignet, dass sich die in einer syndromalen Taubheitsform,
erst von sich nicht überlappenden Re Tektorialmembran schon zwischen der dem BranchioOtoRenalenSyndrom,
gionen ausgegangen worden ist. Es zwölften und zwanzigsten Woche der mutiert. EYA4 kartiert in den DFNA10
handelte sich in allen Familien um eine Embryonalentwicklung ausbildet. Eine Lokus und war deshalb ein gutes Kan
Tieftonschwerhörigkeit. Mutationen Störung der Entwicklung der Tekto didatengen für diese Form der ADNS
konnten erst kürzlich im sog. Wolfra rialmembran, wie sie z.B. durch die HL, zumal es im sich entwickelnden
minGen (WFS1) gefunden werden beschriebenen Mutationen zu erwar Ohr exprimiert ist. Tatsächlich fanden
(Bespalova, 2001; Young, 2001), wel ten ist, würde somit schon zu einem sich in den DFNA10Familien Mutatio
ches auch im autosomal rezessiven sehr frühen Zeitpunkt die Hörfähigkeit nen im EYA4Gen, die zu einem Pro
WolframSyndrom mutiert ist (Strom, stark beeinträchtigen und zu einem teinabbruch führen (Wayne, 2001). Da
1998). Dieses Syndrom ist durch das prälingualen Auftreten der Hörstörung die Hörstörung in diesen Familien spät
Vorliegen eines Diabetes insipidus, führen. manifest ist, scheint dieser transkrip
Diabetes mellitus, einer Optikusatro tionelle Aktivator nicht nur für die Ent
phie und Hörstörungen (deshalb auch DFNA9 – ADNSHL und die wicklung des Innenohrs, sondern
als DIDMOAD bezeichnet) charakteri Menièrsche Erkrankung? auch für die Aufrechterhaltung der
siert, zusätzlich kann es zu weiteren, Durch die Analyse innenohrspezifi Innenohrstrukturen im späteren Leben
oft neuropsychiatrischen Symptomen scher Gene konnte ein neues, verantwortlich zu sein.
kommen. Bis jetzt ist nicht bekannt, kochleär exprimiertes Gen identifiziert
dass Anlageträger für das Wolfram werden, das COCH genannt wurde. DFNA11 – NSHL und das Usher
Syndrom ein erhöhtes Risiko für das Dem Protein konnte durch Sequenz Syndrom
Auftreten einer Hörstörung haben. In vergleiche keine Funktion zugeordnet Unkonventionelle Myosine (d.h. Nicht
teressanterweise liegen alle Mutatio werden, wenngleich Domänen im Pro Muskel Myosine) sind im Innenohr ex
nen bei der ADNSHL im Cterminalen tein zu finden sind, die auch in schon primiert und wahrscheinlich an einer
Bereich des Gens, wenngleich es für bekannten Proteinen vorkommen. In Vielzahl von Funktionen, wie z.B. der
eine GenotypPhänotyp Korrelation si drei miteinander nicht verwandten Strukturbildung im Bereich der Ste
cherlich noch zu früh ist. DFNA9Familien wurden Mutationen reozilien oder dem Vesikeltransport in
im COCH gefunden (Robertson, Haarzellen, beteiligt, so dass es nicht
DFNA8/12 – Eine Störung im 1998). In allen drei Familien waren erstaunlich war, dass Mitglieder dieser
Aufbau der Tektorialmembran schon vorher histopathologische Ver Genfamilie bei Hörstörungen mutiert
Die Tektorialmembran ist eine azel änderungen mit Ablagerung einer azi sind. Das erste für eine Hörstörung
luläre Membran, die die Haarzellen dophilen Grundsubstanz im Innenohr identifizierte Myosin war das MYO7A,
des Innenohrs bedeckt und für die beschrieben worden. Die Expression welches auch im UsherSyndrom
mechanoelektrische Signaltransdukti von COCH lokalisierte dabei genau in (Taubheit und Retinitis pigmentosa)
on notwendig ist. Die Membran be den Regionen, in denen diese Ablage und bei der autosomal rezessiven
steht aus verschiedenen Kollagenen rungen gefunden werden konnten, so Taubheit mutiert ist (s. a. entspre
und den Tektorinen. Da das humane dass ein ursächlicher Zusammenhang chende Beiträge in diesem Schwer
αTektorin (TECTA) in einer Region zwischen Ablagerung von Mukopoly punkt). Bei der ADNSHL fand sich
von Chromosom 11 liegt, in die auch sacchariden und einem Funktionsaus eine dominante Mutation im Bereich
zwei ADNSHLLoci (DFNA8 und fall von COCH wahrscheinlich ist. So des MYO7AProteins, der zwischen
DFNA12) kartiert wurden, war es ein mit scheint COCH für die strukurelle Kopf und Schwanzdomäne liegt und
perfektes Kandidatengen für die AD Integrität des Innenohrs von Bedeu dem eine Dimerisierungsfunktion zu
NSHL. Tatsächlich konnten Mutatio tung zu sein und eventuell mit Protei geschrieben wird (Liu, 1997). Kann es
nen in beiden Familien gefunden wer nen der Extrazellulärmatrix zu inter durch die Veränderung zu keiner ef
den (Verhoeven, 1998). Beide Muta agieren. Interessanterweise scheinen fektiven Zusammenlagerung von zwei
tionen befinden sich in einem Protein Träger der Mutation im COCHGen ein Myosinen mehr kommen, werden
abschnitt, der sog. „Zona pellucida“ erhöhtes Risiko für das Auftreten der durch die Mutation auch nicht mutier
Domäne, der an ProteinProteinInter Menièrschen Erkrankung zu besitzen te Myosine in ihrer Funktion behindert,
aktionen beteiligt und evolutionär (Fransen, 1999), in der es neben dem d.h. es liegt ein dominantnegativer
hoch konserviert ist. Durch die Verän Hörverlust noch zu Schwindel und Effekt vor. Ähnlich wie bei Mutationen
derungen in diesem funktionell wich Tinnitus kommt. Ob Mutationen im in Connexinen oder Kaliumkanälen
tigen Proteinbereich kommt es wahr COCHGen auch für sporadische Fäl führt dieser dominantnegative Effekt
scheinlich zu Störungen der Protein zum Funktionsverlust von weit über
34 medgen 14 (2002)Genetik der HNO Krankheiten
50% und somit zum dominanten Erb nismen letztlich verstanden sind. Ne entwickeln zu können. Glücklicher
gang. ben der wahrscheinlich selteneren Be weise gibt es wirkungsvolle apparati
teiligung an der ADNSHL (DFNA17, ve Therapieoptionen bei der ADNSHL.
DFNA13 – ADNSHL und das (Lalwani, 2000)) sind Mutationen im Dennoch liegt im besseren Verständ
Stickler Syndrom MYH9 außerdem bei der MayHegglin nis der Embryologie, der Anatomie
Ein weiteres Beispiel für die Allelie von Anomalie, dem FechtnerSyndrom, und der Physiologie der Schlüssel zu
syndromalen und nichtsyndromalen dem SebastianSyndrom, dem Ep einer optimierten medizinischen Be
Hörstörungen zeigt sich bei Mutatio steinSyndrom und dem AlportSyn treuung und Versorgung. Dies gilt, ob
nen im Kollagen11A2Gen. Diese sind drom mit Makrothrombozytopenie be wohl die meisten Formen der ADNS
sowohl für DFNA13 (McGuirt, 1999) schrieben (Heath, 2001), wobei diese HL selten sind, insbesondere deswe
als auch für die nichtokuläre Form Syndrome im wesentlichen durch cha gen, weil die autosomal dominanten
des SticklerSyndroms verantwortlich rakteristische Auffälligkeiten der „Schwerhörigkeitsgene“ wahrschein
(Vikkula, 1995), welches durch das Thrombozyten und z.T. Vorliegen von lich auch für die Entwicklung häufiger
Vorliegen einer Hörstörung und einer Nierenfunktionsstörungen und Hör Hörstörungen wie den M. Menière und
Osteochondrodysplasie gekennzeich störungen gekennzeichnet sind. die Presbyakusis eine Rolle spielen.
net ist. Die DFNA13Mutationen be
finden sich dabei im Bereich der sog. DFNA22 – „Snell’s Waltzer“ und
„TripleHelixDomäne“ des COL11A2. noch ein Myosin
Ultrastrukturelle Untersuchungen an Im Innenohr ist MYO6, ein weiteres
der Maus haben gezeigt, dass das unkonventionelles Myosin, spezifisch Literatur
Bespalova IN, Van Camp G, Bom SJ, Brown
COL11A2 im Innenohr in der Tekto in den sensorischen Haarzellen lokali DJ, Cryns K, DeWan AT, Erson AE, Flothmann
rialmembran vorkommt und dort eine siert, wobei eine besonders hohe K, Kunst HP, Kurnool P, Sivakumaran TA,
strukturgebende Aufgabe besitzt. Konzentration in der Kutikularplatte zu Cremers CW, Leal SM, Burmeister M,
Lesperance MM (2001) Mutations in the Wolfram
finden ist. Somit scheint MYO6 funk syndrome 1 gene (WFS1) are a common cause of
DFNA15 – Der Transkriptionsfaktor tionell an der Verankerung der Stereo low frequency sensorineural hearing loss. Hum
POU4F3 zilien in der Haarzelle beteiligt zu sein. Mol Genet 10: 25018.
Eine spezielle Subklasse von Trans Durch Mutationen des MYO6 in der Davis AC (1989) The prevalence of hearing
kriptionsfaktoren sind die „POU Maus (sog. Snell´s Waltzer) kommt es impairment and reported hearing disability
Domäne“ Transkriptionsfaktoren, die zur Störung der strukturellen Integrität among adults in Great Britain. Int J Epidemiol 18:
9117.
evolutionär weit verbreitet und an ei der Haarzellen und somit zur Taubheit.
ner Vielzahl unterschiedlicher Diffe Erst kürzlich konnte eine Missense del Castillo I Villamar M MorenoPelayo MA del
renzierungsvorgänge häufig neurona Mutation in einer ADNSHLFamilie Castillo FJ Alvarez A Telleria D Menendez I
Moreno F (2002) A deletion involving the
ler Zelltypen beteiligt sind. Ein Vertre (DFNA22) im humanen MYO6 identifi connexin 30 gene in nonsyndromic hearing
ter dieser Klasse ist der Typ IV POU ziert werden (Melchionda, 2001), die impairment. N Engl J Med 346 2439.
Transkriptionsfaktor3 (POU4F3), der die Bedeutung dieses Myosins für die
Denoyelle F, LinaGranade G, Plauchu H,
sowohl in der Embryonalentwicklung Struktur des Innenohres auch beim Bruzzone R, Chaib H, LeviAcobas F, Weil D,
als auch im adulten Zustand stark in Menschen unterstreicht. Petit C (1998) Connexin 26 gene linked to a
den Haarzellen des Innenohrs und des dominant deafness. Nature 393: 31920.
Vestibularorgans exprimiert ist und im Ausblick Fransen E, Verstreken M, Verhagen WI, Wuyts
KnockoutMausmodel zur Taubheit Zusammenfassend, mit 15 bekannten FL, Huygen PL, D'Haese P, Robertson NG,
führt. Tatsächlich konnte auch beim Genen und mehr als 35 Loci, wissen Morton CC, McGuirt WT, Smith RJ, Declau F,
Van de Heyning PH, Van Camp G (1999) High
Menschen eine 8bp Deletion im wir heute unvergleichlich viel mehr als prevalence of symptoms of Meniere's disease in
POU4F3Gen in einer DFNA15Fami noch vor wenigen Jahren – der erste three families with a mutation in the COCH gene.
lie entdeckt werden (Vahava, 1998). Lokus für die ADNSHL wurde 1992 Hum Mol Genet 8: 14259.
Durch diese Deletion kommt es zu ei kartiert und das erste ADNSHLGen Grifa A, Wagner CA, D'Ambrosio L, Melchionda
ner Trunkation des Proteinprodukts. 1997 gefunden. Wir kennen z.B. die S, Bernardi F, LopezBigas N, Rabionet R,
POU4F3 scheint nicht nur für die in molekulare Identität von Transkripti Arbones M, Monica MD, Estivill X, Zelante L,
Lang F, Gasparini P (1999) Mutations in GJB6
itiale Differenzierung der Haarzellen, onsfaktoren, die für die Innenohrent cause nonsyndromic autosomal dominant
sondern auch für die Aufrechterhal wicklung notwendig sind, von Zyto deafness at DFNA3 locus. Nat Genet 23: 168.
tung des ausdifferenzierten Zustands skelettproteinen und Proteinen der Ex
Heath KE, CamposBarros A, Toren A,
der Haarzellen verantwortlich zu sein, trazellulärmatrix, die für die strukturel RozenfeldGranot G, Carlsson LE, Savige J,
was die späte Krankheitsmanifestati le Integrität und Funktionstüchtigkeit Denison JC, Gregory MC, White JG, Barker DF,
on zeigt (ähnlich wie EYA4). der hochspezialisierten Innenohr Greinacher A, Epstein CJ, Glucksman MJ,
Martignetti JA (2001) Nonmuscle myosin heavy
strukturen verantwortlich sind, und chain IIA mutations define a spectrum of
DFNA17 – Ein unkonventionelles von Ionentransportproteinen, die für autosomal dominant macrothrombocytopenias:
Myosin und diverse Phänotypen die Aufrechterhaltung der fein regu MayHegglin anomaly and Fechtner Sebastian
Epstein and Alportlike syndromes. Am J Hum
Das unkonventionelle Myosin MYH9 lierten Ionenhomöostase sorgen. Ver Genet 69: 103345.
ist für eine Vielzahl von Phänotypen stehen allerdings tun wir immer noch
verantwortlich, ohne dass die hierfür relativ wenig – zu wenig, um eine spe Kubisch C, Schroeder BC, Friedrich T,
Luetjohann B, ElAmraoui A, Marlin S, Petit C,
verantwortlichen molekularen Mecha zifische Prophylaxe und/oder Therapie Jentsch TJ (1999) KCNQ4 a novel potassium
medgen 14 (2002) 35Genetik der HNO Krankheiten
channel expressed in sensory outer hair cells is Scharfe C, Rabl W, Gerbitz KD, Meitinger T Korrespondenzadresse
mutated in dominant deafness. Cell 96: 437446. (1998) Diabetes insipidus diabetes mellitus optic Dr. med. Christian Kubisch
atrophy and deafness (DIDMOAD) caused by Institut für Humangenetik
Lalwani AK, Goldstein JA, Kelley MJ, Luxford mutations in a novel gene (wolframin) coding for Universitätsklinikum Bonn
W, Castelein CM, Mhatre AN (2000) Human a predicted transmembrane protein. Hum Mol Wilhelmstr. 31
nonsyndromic hereditary deafness DFNA17 is Genet 7: 20218. D53111 Bonn
due to a mutation in nonmuscle myosin MYH9. Tel. +49228287 2342
Am J Hum Genet 67: 11218. Vahava O, Morell R, Lynch ED, Weiss S, Kagan Fax +49228287 2380
ME, Ahituv N, Morrow JE, Lee MK, Skvorak Christian.Kubisch@ukb.unibonn.de
Lamartine J, Munhoz Essenfelder G, Kibar Z, AB, Morton CC, Blumenfeld A, Frydman M,
Lanneluc I, Callouet E, Laoudj D, Lemaitre G, Friedman TB, King MC, Avraham KB (1998)
Hand C, Hayflick SJ, Zonana J, Antonarakis S, Mutation in transcription factor POU4F3
Radhakrishna U, Kelsell DP, Christianson AL, associated with inherited progressive hearing
Pitaval A, Der Kaloustian V, Fraser C, Blanchet loss in humans. Science 279: 19504.
Bardon C, Rouleau GA, Waksman G (2000)
Mutations in GJB6 cause hidrotic ectodermal Van Camp G, Smith RJH (2002) Hereditary
dysplasia. Nat Genet 26: 1424. Hearing Loss Homepage http://dnalabwww.
uia.ac.be/dnalab/hhh
Liu XZ, Walsh J, Tamagawa Y, Kitamura K,
Nishizawa M, Steel KP, Brown SD (1997) Van Hauwe P, Coucke PJ, Declau F, Kunst H,
Autosomal dominant nonsyndromic deafness Ensink RJ, Marres HA, Cremers CW, Djelantik
caused by a mutation in the myosin VIIA gene. B, Smith SD, Kelley P, Van de Heyning PH, Van
Nat Genet 17: 2689. Camp G (1999) Deafness linked to DFNA2: one
locus but how many genes? Nat Genet 21: 263.
Liu XZ, Xia XJ, Xu LR, Pandya A, Liang CY,
Blanton SH, Brown SD, Steel KP, Nance WE Van Laer L, Huizing EH, Verstreken M, van
(2000) Mutations in connexin31 underlie Zuijlen D, Wauters JG, Bossuyt PJ, Van de
recessive as well as dominant nonsyndromic Heyning P, McGuirt WT, Smith RJ, Willems PJ,
hearing loss. Hum Mol Genet 9: 637. Legan PK, Richardson GP, Van Camp G (1998)
Nonsyndromic hearing impairment is associated
Lynch ED, Lee MK, Morrow JE, Welcsh PL, with a mutation in DFNA5. Nat Genet 20: 1947.
Leon PE, King MC (1997) Nonsyndromic
deafness DFNA1 associated with mutation of a Verhoeven K, Van Laer L, Kirschhofer K, Legan
human homolog of the Drosophila gene PK, Hughes DC, Schatteman I, Verstreken M,
diaphanous. Science 278: 13158. Van Hauwe P, Coucke P, Chen A, Smith RJ,
Somers T, Offeciers FE, Van de Heyning P,
Marazita ML, Ploughman LM, Rawlings B, Richardson GP, Wachtler F, Kimberling WJ,
Remington E, Arnos KS, Nance WE (1993) Willems PJ, Govaerts PJ, Van Camp G (1998)
Genetic epidemiological studies of earlyonset Mutations in the human alphatectorin gene
deafness in the U.S. schoolage population. Am cause autosomal dominant non syndromic
J Med Genet 46: 48691. hearing impairment. Nat Genet 19: 602.
McGuirt WT, Prasad SD, Griffith AJ, Kunst HP, Vikkula M, Mariman EC, Lui VC, Zhidkova NI,
Green GE, Shpargel KB, Runge C, Huybrechts Tiller GE, Goldring MB, van Beersum SE, de
C, Mueller RF, Lynch E, King MC, Brunner HG, Waal Malefijt MC, van den Hoogen FH, Ropers
Cremers CW, Takanosu M, Li SW, Arita M, HH, und et al (1995) Autosomal dominant and
Mayne R, Prockop DJ, Van Camp G, Smith RJ recessive osteochondrodysplasias associated
(1999) Mutations in COL11A2 cause non with the COL11A2 locus. Cell 80: 4317.
syndromic hearing loss (DFNA13). Nat Genet 23:
4139. Wayne S, Robertson NG, DeClau F, Chen N,
Verhoeven K, Prasad S, Tranebjarg L, Morton
Melchionda S, Ahituv N, Bisceglia L, Sobe T, CC, Ryan AF, Van Camp G, Smith RJ (2001)
Glaser F, Rabionet R, Arbones ML, Notarangelo Mutations in the transcriptional activator EYA4
A, Di Iorio E, Carella M, Zelante L, Estivill X, cause lateonset deafness at the DFNA10 locus.
Avraham KB, Gasparini P (2001) MYO6 the Hum Mol Genet 10: 195200.
human homologue of the gene responsible for
deafness in Snell's waltzer mice is mutated in Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai
autosomal dominant nonsyndromic hearing loss. HP, Zhang BR, Xie W, Hu DX, Zheng D, Shi
Am J Hum Genet 69: 63540. XL, Wang DA, Xia K, Yu KP, Liao XD, Feng Y,
Yang YF, Xiao JY, Xie DH, Huang JZ (1998)
Morton NE (1991) Genetic epidemiology of Mutations in the gene encoding gap junction pro
hearing impairment. Ann N Y Acad Sci 630: 16 tein beta3 associated with autosomal dominant
31. hearing impairment. Nat Genet 20: 3703.
Richard G, Smith LE, Bailey RA, Itin P, Hohl D, Young TL, Ives E, Lynch E, Person R, Snook S,
Epstein EH, Jr, DiGiovanna JJ, Compton JG, MacLaren L, Cator T, Griffin A, Fernandez B,
Bale SJ (1998) Mutations in the human connexin Lee MK, King MC (2001) Nonsyndromic
gene GJB3 cause erythrokeratodermia variabilis. progressive hearing loss DFNA38 is caused by
Nat Genet 20: 3669. heterozygous missense mutation in the Wolfram
syndrome gene WFS1. Hum Mol Genet 10: 2509
Robertson NG, Lu L, Heller S, Merchant SN, 14.
Eavey RD, McKenna M, Nadol JB, Jr,
Miyamoto RT, Linthicum FH, Jr, Lubianca Neto
JF, Hudspeth AJ, Seidman CE, Morton CC,
Seidman JG (1998) Mutations in a novel cochlear
gene cause DFNA9 a human nonsyndromic
deafness with vestibular dysfunction. Nat Genet
20: 299303.
Strom TM, Hörtnagel K, Hofmann S, Gekeler F,
36 medgen 14 (2002)Sie können auch lesen

















































