Früh- und Differentialdiagnose der Parkinson-Syndrome - Thomas Gasser Zentrum für Neurologie der Universität
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Früh- und Differentialdiagnose der
Parkinson-Syndrome
Thomas Gasser
Zentrum für Neurologie der Universität
Tübingen
Neurologische Klinik und Hertie-Institut für
Klinische Hirnforschung
Abteilung für Neurologie mit Schwerpunkt
Neurodegeneration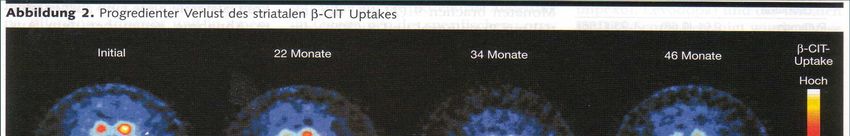
State of the Art
• Klinische Diagnose des Parkinson-Syndroms
– 2 von 4 Kardinalsymptomen
• Akinese, Rigor, Ruhetremor, posturale Störung
• Ausschluss eines sekundären PS
– Strukturelle Bildgebung (NPH, vaskulär)
• Abgrenzung IPS vs. Atypisches PS
– MSA, PSP, CBD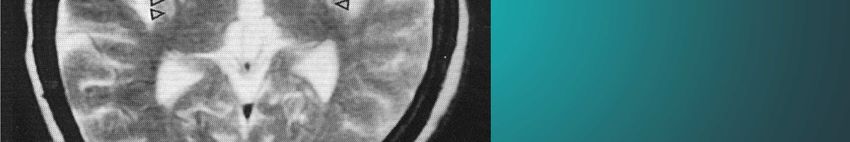
Parkinson-Syndrome • Parkinson-Krankheit 80% – Sporadisch 90% – Familiär 10% • Atypische Parkinson-Syndrome 20% – Multisystematrophie – Progressive Supranukleäre Blickparese – Corticobasale Degeneration • Symptomatische Parkinson-Syndrome – Vaskuläres Parkinson-Syndrom – NPH – andere
Hauptkriterien der British-Brain-Bank für die
klinische Diagnose des IPS (1-3)
1 Diagose eines 2 Diagnose der 3 Ausschlusskriterien für die
Parkinson-Syndroms Parkinson-Krankheit Diagnose der Parkinson-Krankheit
Bradykinesie Erfüllung der Kriterien für 1 Nachweis von mindestens 1
plus mindestens 1 plus mindestens 3 der der folgenden Kriterien:
der folgenden Symptome: folgenden Kriterien:
• wiederholte zerebrale Ischämien
• Rigor • einseitiger Beginn • wiederholte SHTs
• 4-7 Hz Ruhetremor • persistierende Asymmetrie • wiederholte Enzephalitiden
• Haltungsinstabilität • Ruhetremor • intrakranielles Neoplasma
• progredienter Verlauf • schubförmige Symptomzunahme
• initial gute L-Dopa-Response • okulogyre Krisen
• Wirksamkeit von L-Dopa • Neuroleptikaeinnahme
> 5 Jahre • Parkinson-Syndrom bei > 1
• >10-jähriger klinischer Verlauf Blutsverwandten
• L-Dopa-getriggerte Chorea • dauerhafte Remission
• strikte Halbseitigkeit > 3 Jahre
• supranukleäre Störung der
Okulomotorik
• zerebelläre Symptomatik
• frühzeitig autonome Störungen
• frühzeitige Demenz
• Pyramidenbahnzeichen
• initial fehlende L-Dopa-Wirksamkeit
Kap. 6.2.7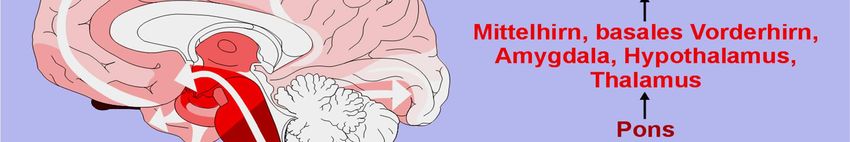
Braak et al., Neurbiol Aging

Veränderung der Riechfunktion
Prämotorisches Parkinson-Syndrom? Video TG1

RBD bei M. Parkinson • 15 – 47 % der Patienten (Comella `98; Gagnon et al. `02) • Bei 52 % trat RBD vor MP auf (Olson et al. `00) • Remissionen sind selten • Auch in anderen neurodegenerativen Erkrankungen (DLB, MSA , SCA 3)
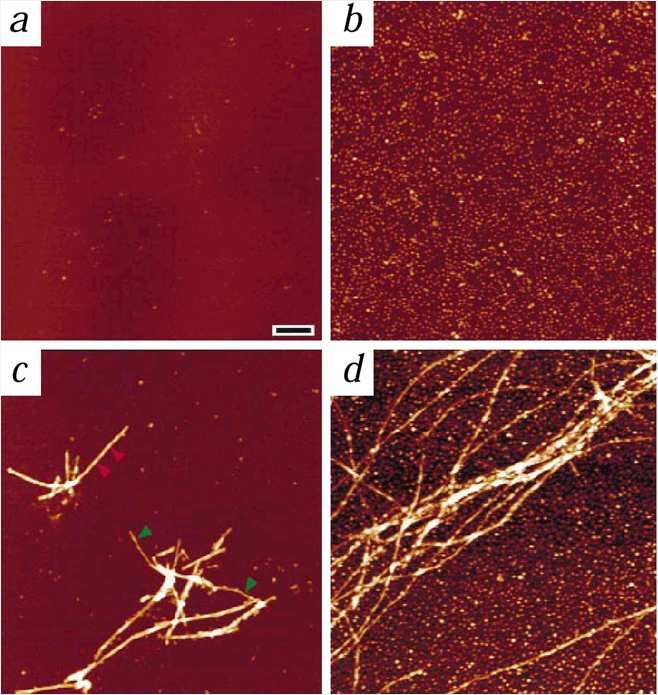
Echogene Substantia nigra
Kontrolle Morbus Parkinson
*
Hyperechogenität der Substantia nigra in gesunden
Kontrollen: MRT Veränderungen
7 Parkinson Patienten
14 Kontrollen (je 7 Kontrollen mit und ohne Hyperechogenität der Substantia nigra)
Kap. 7.2.8 Behnke et al., Neuroimage 2009; 47: 1237-1243Hyperechogenität der Substantia nigra in gesunden
Kontrollen: dopaminerger Zellverlust (F-Dopa PET)
7 Parkinson Patienten
14 Kontrollen (je 7 Kontrollen mit und ohne Hyperechogenität der Substantia nigra)
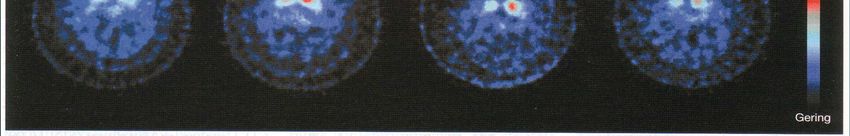
Kap. 7.2.9 Behnke et al., Neuroimage 2009; 47: 1237-1243IPS und ß-CIT-SPECT
Reduktion ß-CIT-Uptake und PD-Progression über 4 Jahre
Kap. 7.3.5Parkinson-Syndrome • Parkinson-Krankheit 80% – Sporadisch 90% – Familiär 10% • Atypische Parkinson-Syndrome 20% – Multisystematrophie – Progressive Supranukleäre Blickparese – Corticobasale Degeneration • Symptomatische Parkinson-Syndrome – Vaskuläres Parkinson-Syndrom – NPH – andere
α-Synuclein-A53T Mutation
Wildtyp
Video TG2 Früher Beginn
Dominante FA
Akinetisch-rigide
mutiert
Häufig Demenz• LRRK2 Mutation
• Dominante FA, oft unvollständige
Penetranz
• Typisches Erkrankungsalter (59 J)
• Typische IPS
• Eher etwas milderer Verlauf
Video TG3Rezessive früh beginnende Parkinson-Syndrome
Parkin > 40 Mutationen
75% bei onset < 20
25% bei onset < 30
5 – 10% bei
onset < 40
PINK1 < 10 Mutationen
1 – 2 % des early-onset
PS
DJ-1 4 Mutationen
seltenGenetik des Parkinson-Syndroms
Recessive genes
LRRK2 GBA
Parkin, PINK-1, DJ1
α-synuclein
unexplained and tau
RisikovariantenMultisystematrophie (MSA)
Klinische Leitsymptomatik Neuropathologie
- Parkinsonismus plus - Vorwiegend in Oligodendro-
• Dysarthrie zyten lokalisierte argyrophile,
• Dysphagie
• autonome Störungen alpha-Synuclein-positive,
(Shy-Drager-Variante) fibrilläre Einschlusskörperchen
• ausgeprägter Antecollis
- Verbreiteter Neuronenverlust
• laryngealer Stridor
• spasmod. Dysphonie und Gliose u. a. in:
• zerebelläre Ataxie • Substantia nigra
(OPCA-Form) • Putamen
• (Demenz)*
• autonomen Kerngebieten •
- schlechte L-Dopa-Response Olive
- rasche Progression
*Die Angaben zur Häufigkeit der Demenz
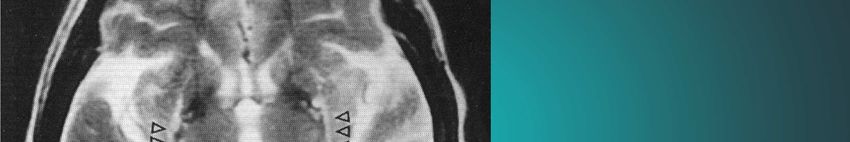
Kap. 6.3.2 sind in der Literatur unterschiedlichAntecollis bei MSA
Quinn N: Disproportionate antecollis in multiple system atrophy.
Kap. 6.3.3
Lancet 1: 844, 1989Red flags bei MSA
• Antecollis
• Myoklonischer Halte- und
Aktionstremor
• REM-Schlaf-Störung
• Respiratorischer Stridor
• Spontane Dystonie (orofacial u.
Platysma) → „Risus sardonicus“
Kap. 6.3.4 Wenning et al., 2003Multisystematrophie cMRT (OPCA-Typus) Deutliche Atrophie von Pons, Medulla und Cerebellum Kap. 7.5.3 mit Erweiterung des IV. Ventrikels
MSA
(SND < OPCA)
„Hot cross bun“
2 Jahre 64%
4 Jahre 100%
Watanabe et al., 2002MSA
(SND > OPCA )
„Hyperintense
putaminal rim“
2 Jahre 38 %
4 Jahre 80 %
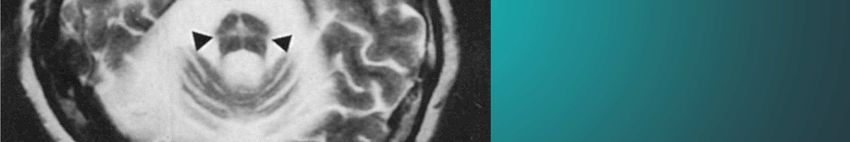
Watanabe et al., 2002„Der erstaunte Blick“ bei PSP Kap. 6.4.1
Progressive supranukleäre Blickparese (PSP)
Klinische Leitsymptomatik Neuropathologie
- Parkinsonismus plus - Atrophie von Pallidum,
• frühzeitige Gangstörung Mittelhirn und Pons
• vertikale Blickparese
• Dysphagie - Abblassung der S. nigra
• Demenz Zellverlust und Gliose u. a. in
• Reklination des Kopfes Pallidum, S. nigra, Nucleus
subthalamicus, Pons
- sehr schlechte bzw. keine
L-Dopa-Response - tau-positive argyrophile
Neurofibrillen in Basal-
- rasche Progression
ganglien und Hirnstamm
Kap. 6.4.3Unterschiedliche klinische Phänotypen bei
pathologisch bestätigter PSP
Klinikopathologische Studie bei 103 Fällen mit gesicherter PSP. Dabei fand sich in einem Drittel
der Fälle eine spezifische, vom üblichen klinischen Bild (Richardson-Syndrom) abweichende
Präsentation (PSP-Parkinsonismus).
„Richardson‘s Syndrom“ PSP-Parkinsonismus
(54% der Fälle) (32% der Fälle)
Früh im Krankheitsverlauf Stürze Asymmetrischer Beginn
Supranukleäre Blickparese Tremor
Kognitive Störung Initial mäßiges Ansprechen auf L-Dopa
M/F-ratio 2/1 M/F-ratio 1/1
4-repeat/3-repeat tau-ratio: 2,84 4-repeat/3-repeat tau-ratio: 1,63
14 % der Fälle konnten keiner spezifischen klinischen Form zugeordnet werden.
Kap. 6.4.4 Williams et al.: Brain 2005;128:1247-1258PSP- 3 klinische Varianten
Richardson’s Syndrom PSP-P PSP-PAGF
axial deutlich mehr als axial weniger oder gleich wie
Rigidität axial
Extr. Extr.
Bradykinesie mild moderat moderat
ja/nein (Ruhe oder „jerky“
Tremor nein nein
postural))
Frühe Stürze ja nein nein
Frühe posturale Instabilität ja nein ja
Frühe kognitive Defizite häufig nein nein
Frühe okulomotorische St. ja nein nein
Dopa-Responsivität nein oft nein
Hyposmie nein nein -
Kardiales MIBG-Szinti. normal normal* normal*
Kap. 6.4.12 Williams and Lees; Lancet Neurol 2009: 8; 270–279 mit ModifikationencMRT bei PSP Verschmächtigung des rostralen Mittelhirns: Kolibri-Zeichen Kap. 7.5.6
MRT eines Patienten mit PSP: Abflachung der
Vierhügelplatte
Quelle:
Peter
Kap. 7.5.7 Vieregge,
LemgoKortikobasale Degeneration (CBD)
Klinische Leitsymptomatik Neuropathologie
- halbseitiger - asymmetrische
Parkinsonismus frontoparietale Atrophie
plus
- achromatische Neuronen-
• Dyspraxie
Schwellung
• kortikal sensor. Defizite
- Zellverlust und Gliose, u. a.
• sog. Alien-Hand-Syndrom in Kortex und S. nigra
• Myoklonus - Tau-positive
(dysprakt. Gangstörung) Einschlusskörperchen
• Demenz
- keine L-Dopa-Response
- rasche Progression
Kap. 6.5.1Kortikobasale Degeneration:
klinische Symptomatik (n=147)
Parkinsonismus
Rigor
Bradykinese
Gangstörung
Tremor
andere mot.Symptome
Dystonie
Myoklonus
kortikale Störung
Dyspraxie
alien limb
Demenz
Aphasie
Verschiedenes
Pyramidenbahnzeichen
Blickparese
Dysarthrie
cerebellär
Schmerzen %
Kap. 6.5.2 0 10 20 30 40 50 60 70 80 90 100
Kompoliti et al., 1998Kortikobasale Degeneration (CBD)
Einschluss Ausschluss
Lang et al., Rigor + 1 kortikales S. (Apraxie, alien • Frühe Demenz
1994 limb) • Frühe vertikale
oder Blickparese
Asymmetrischer Rigor, Dystonie u. • Ruhetremor
Reflexmyoklonus
• Deutliche
Kumar et al., Progredienter Verlauf, asymmetrischer
Beginn, kortikale Störung autonome Störung
1998
• Ansprechen auf Dopa
und
Akinetisch-rigides Syndrom, Dopa-
resistent, Dystonie, fokaler Myoklonus
Kap. 6.5.3 Movement Disorders Society Scientific Issues Committee Report: Litvan et al., 2003Kortikobasale Degeneration (CBD)
cMRT
Unilaterale kortikale Atrophie der Perizentralregion
(Nachweisbarkeit
Kap. 7.5.8
in >50 % der Fälle)Atypische Parkinson-Syndrome:
klinische Differentialdiagnose
MSA
Frühzeitig schwere autonome Störungen
(orthostatische Hypotension, Synkopen, Impotenz,
Urininkontinenz oder -retention, Anhidrose)
Antecollis
Deutliche Dysarthrie oder Dysphagie
Inspiratorischer Stridor
Irregulärer Haltetremor
PSP
Supranukleäre vertikale Blickparese
Frühe posturale Instabilität und Stürze
Zeichen der frontalen Disinhibition (Applauszeichen)
CBD
Starke Asymmetrie der Symptome
Einseitige ideomotorische Apraxie
„Alien limb“ Phänomen (nur bei 50 %)Atypische Parkinson-Syndrome
demaskieren sich in den ersten 4 Jahren
Orthostatische Hypotension
MSA
Stürze
MSA Dysarthrie
Dysarthrie CBD
PSP, MSA
Dysarthrie
Urininkontinenz DLB
MSA
Stürze Stürze
Stürze
CBD DLB
PSP
0 5 10 15 20 25 30 35 40 45 50
Monate Lachenmayer: J Neurol 2003
Kap. 6.1.3Retrocollis M. Parkinson Antecollis PSP MSA Kap. 6.1.4
SWEDDS
Video TG6F-Dopa PET
Patienten im Video:
Scans without
evidence of
dopaminergic
deficit (SWEDDs)Sie können auch lesen

















































