Analyse des Repertoires der variablen Region von Immunglobulinen und Detektion Desmoglein-3-spezifischer B-Zellen im peripheren Blut von Patienten ...
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

Aus der Klinik für Dermatologie, Allergologie und Venerologie der Universität zu Lübeck Direktor: Prof. Dr. D. Zillikens Analyse des Repertoires der variablen Region von Immunglobulinen und Detektion Desmoglein-3-spezifischer B-Zellen im peripheren Blut von Patienten mit Pemphigus vulgaris Inauguraldissertation zur Erlangung der Doktorwürde der Universität zu Lübeck - aus der Sektion Medizin - Vorgelegt von: Christoph Arolt aus Lübeck Lübeck 2019
1. Berichterstatter: Priv.-Doz. Dr. Andreas Recke 2. Berichterstatter: Priv.-Doz. Dr. Niklas Gebauer Tag der mündlichen Prüfung: 18.06.2020 Zum Druck genehmigt. Lübeck, den 18.06.2020 - Promotionskommission der Sektion Medizin -
Inhaltsverzeichnis Inhaltsverzeichnis ................................................................................................................... I Abkürzungsverzeichnis ...................................................................................................... III 1. Einleitung und Fragestellung .......................................................................................... 1 1.1. Pemphigus vulgaris ......................................................................................................... 1 1.2. Pathogenität von Pemphigus vulgaris-AAK ............................................................... 3 1.3. Selbsttoleranz und ihre Schwachstellen in der B-Zell-Entwicklung ........................ 6 1.4. Genetische Charakteristika der Anti-Dsg3-Immunglobuline ................................. 11 1.5. Desmoglein-3 spezifische B-Zellen .............................................................................. 12 1.6. BZR-Repertoires ............................................................................................................. 14 1.7. Fragestellung .................................................................................................................. 16 2. Material und Methoden .................................................................................................. 17 2.1. Material ............................................................................................................................ 17 2.1.1. Geräte und Laborbedarf ............................................................................................. 17 2.1.2. Puffer, Medien und Lösungen ................................................................................... 19 2.1.3. Chemikalien................................................................................................................ 20 2.1.4. Kits und Enzyme ....................................................................................................... 20 2.1.5. Antikörper, Konjugatfarbstoffe, Kompensationshilfen .............................................. 21 2.1.6. Ethischer und datenschutzrechtlicher Umgang mit Patientenmaterialien ............... 22 2.2. Methoden ........................................................................................................................ 23 2.2.1. Herstellung von Desmoglein-3-Dylight650 .............................................................. 23 2.2.2. Desmoglein-3-spezifischer ELISA ............................................................................. 23 2.2.3. Durchflusszytometrische Evaluation des Desmoglein-3-Dylight650 ....................... 24 2.2.4. Vorbereitung humanen Vollblutes für die durchflusszytometrische Untersuchung 25 2.2.5. DNA-Extraktion, Amplifizierung per PCR und Sequenzierung der schweren Kette der B-Zell-Subpopulationen ................................................................. 26 2.2.6. Statistische Analyse ................................................................................................... 27 2.2.7. Definition von Klonen und Bestimmung der Klon-Größe ........................................ 28 2.2.8. Analyse der Verteilung und Variabilität der VH- und DH-Genbenutzung ............... 28 3. Ergebnisse ......................................................................................................................... 30 3.1. Entwicklung einer durchflusszytometrischen Methode zur Analyse von B-Zellen .................................................................................................... 30 3.2. Fluoreszenz-Markierung von Desmoglein-3 ............................................................. 33 3.3. Bei Konjugation von Desmoglein-3 mit Dylight650 bleibt die Reaktivität mit PV-Seren erhalten........................................................................................................... 34 3.4. Die Konjugation von Desmoglein-3 und Dylight650 lässt sich durchflusszytometrisch nachweisen .................................................................................. 36 3.5. Das entworfene durchflusszytometrische Panel kann unterschiedliche B-Zell-Subpopulationen diskriminieren ............................................................................ 39 I
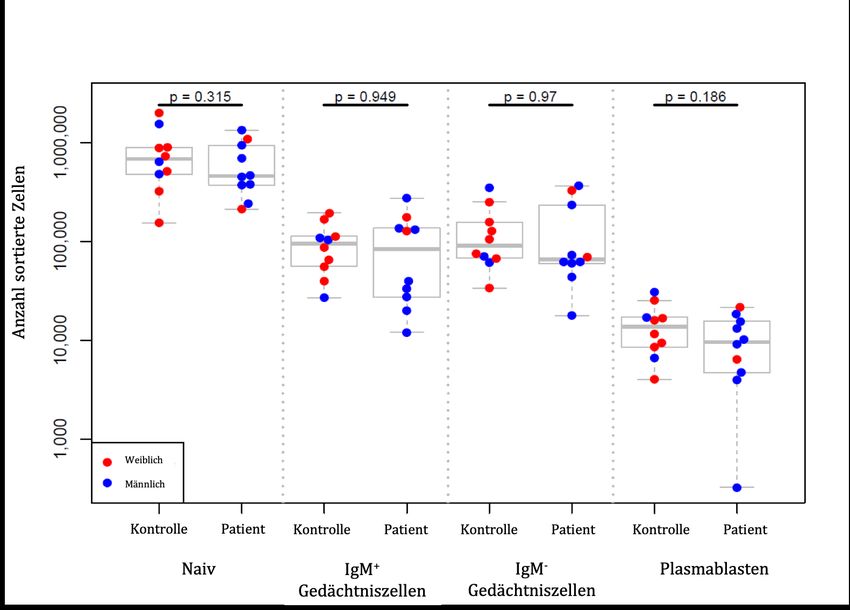
3.6. Zwischen PV-Patienten und Kontrollen bestehen keine signifikanten Unterschiede hinsichtlich der relativen Größe der B-Zell-Subpopulationen............... 41 3.7. Detektion Desmoglein-3-spezifischer B-Zellen in der IgM-- Gedächtniszellpopulation .................................................................................................... 45 3.8. Sequenzen im BZR-Repertoire von PV-Patienten verteilen sich in weniger, dafür größeren Klongruppen als Kontrollen .................................................................... 48 3.9. Die Analyse der VH- und DH-Gen-Benutzung zeigt keine gerichteten Unterschiede zwischen PV-Patienten und gesunden Kontrollen .................................. 53 3.10. Ein bislang mit PV-AK assoziiertes Aminosäuremotiv ist bei Patienten-BZR nicht häufiger kodiert als bei Kontrollen ................................................ 54 4. Diskussion......................................................................................................................... 55 4.1. Entwicklung und Evaluation einer Methode zur Detektion autoreaktiver B-Zellen mittels Durchflusszytometrie .............................................................................. 55 4.2. Entwicklung und Evaluation einer Methode zur Trennung von B-Zellen in vier Subpopulationen....................................................................................................... 59 4.3. Untersuchungen zum B-Zell-Rezeptor-Repertoire bei Pemphigus vulgaris ........ 60 4.3.1. Rekrutierung von Patienten und Kontrollen ............................................................ 61 4.3.2. Probenaufarbeitung ................................................................................................... 63 4.3.3. DNA-Reinigung und PCR-Amplifikationsstrategie ................................................. 64 4.3.4. Datenanalyse.............................................................................................................. 66 4.4. Schlussfolgerungen ........................................................................................................ 69 5. Zusammenfassung ........................................................................................................... 71 6. Literaturverzeichnis......................................................................................................... 72 7. Abbildungs- und Tabellenverzeichnis ........................................................................ 81 8. Danksagung ...................................................................................................................... 83 9. Lebenslauf ......................................................................................................................... 85 II
Abkürzungsverzeichnis Abkürzung Bedeutung Abb. Abbildung AIBD Autoimmune blistering disease AK Antikörper AAK Autoantikörper AS Aminosäure BZR B-Zell-Rezeptor BZ B-Zelle, von Bursa Fabricii CD Cluster of differentiation CDR Complement determining region CR Komplette Remission DNA Desoxyribonukleinsäure Dsg1 Desmoglein-1 Dsg3 Desmoglein-3 ELISA Enzyme linked immunosorbent assay FACS Fluorescence activated cell sorting FSC-A Forward light scatter – area FSC-H Forward light scatter – heigth HLA Human Leukocyte Antigen Ig Immunglobulin IgG Immunglobulin G IgM Immunglobulin M IL Interleukin IR Inkomplette Remission KM Knochenmark NGS Next generation sequencing OD Optische Dichte PB Plasmablast PBMC Peripheral blood mononuclear cells PCR Polymerase chain reaction PV Pemphigus vulgaris R-Mutation Replacement mutation RA Rheumatoide Arthritis rpm Umdrehungen pro minute RT Raumtemperatur, 22 °C S-Mutation Silent mutation SHM Somatische Hypermutation SLE Systemischer Lupus erythematodes Tab. Tabelle TBS Tris-buffered salin III
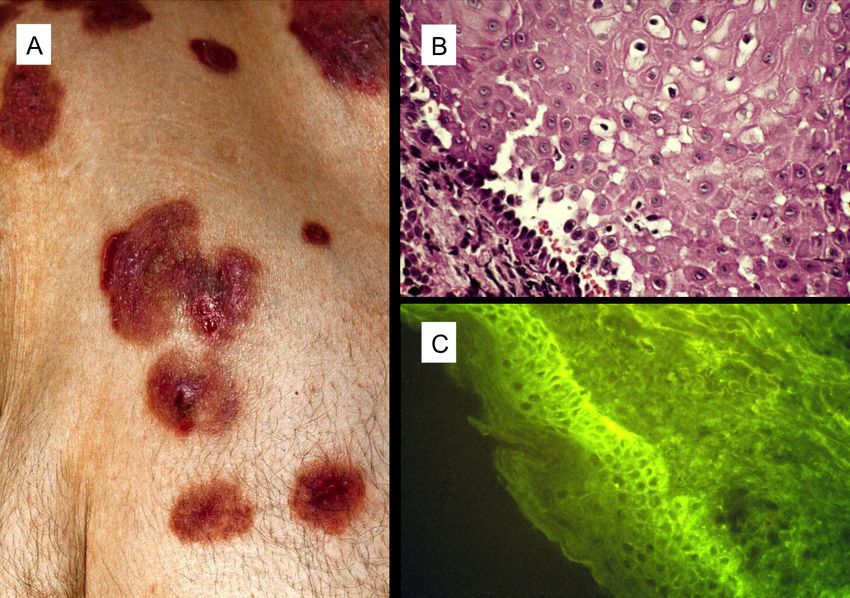
1. Einleitung und Fragestellung 1.1. Pemphigus vulgaris Pemphigus vulgaris (PV) ist eine durch anti-Desmoglein-3-Antikörper vermittelte, blasenbildende Autoimmundermatose (autoimmune blistering disease, AIBD), welche chronisch verläuft und unbehandelt letal enden kann. Sie ist mit einer Inzidenz von 94 / 1.000.000 Einwohner und Jahr eine der häufigsten blasenbildenden Autoimmundermatosen in Deutschland (1), wobei Frauen (2), ebenso wie Ashkenasim häufiger erkranken (3). Die Erkrankung tritt meist in der vierten bis sechsten Lebensdekade auf (4). Eine Studie von Langan et al. ergab für Patienten mit PV unter Therapie eine dreifach höhere Gesamtmortalität im Vergleich zu gesunden Kontrollen (2). Wichtigstes klinisches Merkmal der Krankheit sind muköse Erosionen, vor allem der Mundschleimhaut, welche durch Ruptur von Blasen entstehen, die häufig bereits nicht mehr beobachtet werden können. Bei etwa der Hälfte der Patienten entstehen weiterhin schlaffe, subepidermale, mit klarem Sekret gefüllte Basen der Epidermis (Abb. 1A). Diese können bei aktiver Erkrankung durch Scherung auf der Haut provoziert werden (Nikolski Phänomen I). Bei bestehenden Blasen kann der Blaseninhalt durch seitlichen Druck verschoben werden (Nikolski Phänomen II). Charakteristisch ist das Vorkommen von Autoantikörpern (AAK) gegen Desmoglein- 3 (Dsg3) beim mukösen und zusätzlich Desmoglein-1 (Dsg1) beim mukokutanen Typ im Serum der Patienten (5). Desmogleine gehören zur Gruppe der Cadherine, sind ein Bestandteil der Desmosomen und spielen somit eine wichtige Rolle bei der intraepidermalen Zell-Zell-Adhäsion (6). In der betroffenen Haut von PV Patienten können AAK mittels direkter Immunfluoreszenz nachgewiesen werden. Die Darstellung von im Blut zirkulierenden Autoantikörpern erfolgt unter anderem anhand der indirekten Immunfluoreszenz auf Affenösophagus-Präparaten (Abb. 1C). Bei beiden 1
Untersuchungen werden AAK-Depositionen mit Hilfe eines fluoreszenzmarkierten anti-IgG-AK sichtbar gemacht. Diese zeigen sich als Fluoreszenz an den Zell-Zell- Grenzflächen. Lichtmikroskopisch kann nach dermatohistopathologischen Routinefärbungen, wie z.B. mit Hämathoxylin und Eosin, eine suprabasale Spaltbildung der Epidermis eine Akantholyse festgestellt werden (Abb. 1B) (7). Zur Therapie des PV kommen Kortikosteroide als Erstlinientherapie zum Einsatz. Trotz ihrer guten Wirkung vergehen oft Jahre, bis eine komplette Remission bei einem Teil der Patienten erreicht werden kann (8). Aus diesem Grund, wie auch zur Reduktion der Kortisondosis, werden außerdem verschiedene Immunmodulatoren bzw. -suppressiva wie Mycophenolatmofetil, Azathioprin, Cyclophosphamid, Dapson und Methotrexat verwendet (6). Zahlreiche Studien zeigen, dass der anti- CD20-AK Rituximab bei therapierefraktärem PV wirksam ist. Voll- bzw. Teilremissionen sind unter Anwendung von Rituximab bei 95 % (9) und Vollremission bei bis zu 66-75 % (10) der Patienten, je nach Protokoll möglich. Auch mit der Kombination von Rituximab mit intravenösen Immunglobulinen (11) sowie mit Immunapherese, Mycophenolatmofetil und Kortikosteroiden (12) können bei therapierefraktärem PV gute Heilungsraten erzielt werden. Bei einem kleinen Kollektiv von 5 Patienten kam es durch Rituximab als Erstlinien- und Monotherapie sogar zu einer Vollremissionsrate von 100 % ohne weitere Therapie über 6 Jahre (13). 2
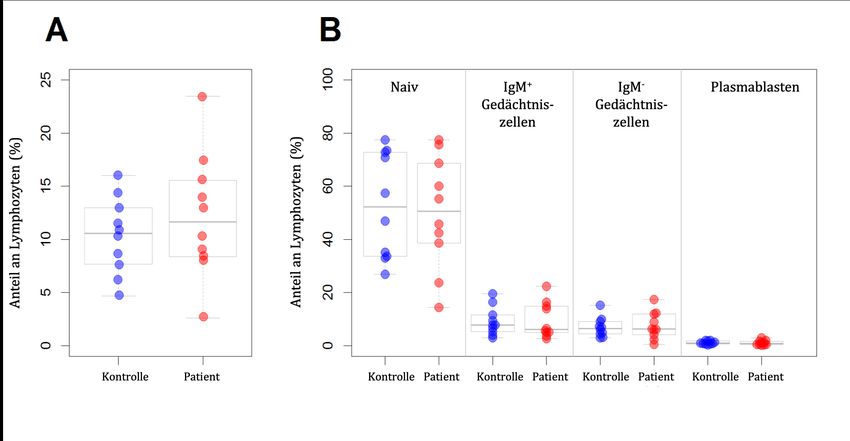
Abbildung 1: Klinisches Bild und mikroskopischer Befund des Pemphigus vulgaris. (A) Schlaffe Blasen auf scheinbar gesunder Haut, die leicht einreißen und Erosionen hinterlassen. (B) Lichtmikroskopisch erkennbare suprabasale Akantholyse (Hämathoxylin- Eosin-Färbung, 200× Vergrößerung). (C) Nachweis einer Interzellulärfluoreszenz zwischen epidermalen Keratinozyten in der indirekten Immunfluoreszenz (Kryoschnitt neonataler Vorhaut, 200× Vergrößerung). 1.2. Pathogenität von Pemphigus vulgaris-AAK Bereits in den späten 1980er und frühen 1990er Jahren wurden die Desmogleine-1 und -3 als PV-Antigene identifiziert, gegen welche sich zirkulierende AAK im Blut von PV Patienten richten (6,14,15). Diese integralen Membranproteine gehören der Familie der Cadherine an und sind Teil der Desmosomen in der Epidermis, welche die Zell- Zell-Adhäsion vermitteln. Cytoplasmatisch ist Desmoglein über Desmoplakin, Plakoglobin und Plakophilin an Intermediärfilamente gebunden. Extrazellulär binden Desmogleine an Desmogleine oder Desmocolline der jeweiligen Nachbarzellen (16). 3
AAK gegen Dsg1 und Dsg3 spielen eine wichtige Rolle in der Pathogenese von PV. Sie sind im Blut von Patienten nachweisbar und zeigten in den meisten Studien eine Korrelation mit der Krankheitsaktivität (13,17–19). Der Nachweis von Anti-Dsg3-AK im ELISA ist sehr spezifisch für PV (20) und essentieller Bestandteil der Diagnostik (7). Die Evidenz, dass anti-Dsg3 AK bei der Pathogenese von PV eine Rolle spielen, folgt aus zahlreichen experimentellen Studien: Mittels der phage display Technik konnten anti-Dsg3-AAK isoliert werden. Ferner zeigte sich, dass sie in humanen Keratinozytenkulturen eine Akantholyse, also eine Spaltbildung in der Epidermis hervorrufen können. Amagai et al. entwickelten ein Protein bestehend aus der extrazellulären Domäne von Dsg3 und der konstanten Region eines Immunglobulins. Sie konnten zeigen, dass Patientensera, deren anti- Dsg3-AAK zuvor durch dieses chimäre Dsg3-Immunglobulin gebunden wurden, ihre Fähigkeit zur Blasenbildung im Mausmodell verlieren (21). Dsg3 Knockout-Mäuse (Dsg -/-), welche mit Dsg3 immunisiert wurden, entwickelten Dsg3-spezifische Splenozyten, welche in immundefizienten Mäusen einen PV-Phänotyp auslösten (22). Weiterhin konnte in einer Studie von Anhalt et al. gezeigt werden, dass aus PV- Patienten isolierte IgG im Mausmodel einen PV Phänotyp induzieren können (23). Experimentell erzeugte AK gegen Dsg3 konnten eine mit PV Präparaten vergleichbare suprabasale Akantholyse im Mausmodel sowie in Keratinozytenkulturen auslösen. Ein aus mit Dsg-3 immunisierten Mausstämmen gewonnener, monoklonaler anti- Dsg3-IgG-AK (AK23) (24) und 3 verschiedene IgG-AK (25), welche aus Patientenseren isoliert wurden, banden an die aminoterminale, extrazelluläre, trans- adhäsive Schnittstelle des Dsg3. Es wird angenommen, dass dies die Adhäsion zweier Dsg3-Moleküle sterisch behindert (24). Diese Inhibition der Desmoglein-Bindung durch sterische Behinderung scheint jedoch nicht die einzige Ursache für die Akantholyse, und für diese allein nicht ausreichend zu sein (26). In Zellkulturen konnte gezeigt werden, dass die Bindung von anti-Dsg3-AK zur Aktivierung eines intrazellulären Signalwegs über p38-mitogenaktivierte Proteinkinasen führt, und so die Reorganisation des Zytoskeletts induziert (27). Die Inhibition dieses Enzyms 4
verhinderte in-vitro die Internalisierung von Dsg3 von der keratinozytären Zelloberfläche (28) und im passiven Mausmodel die Entstehung des PV- Phänotyps (29). Der anti-Dsg3-AAK-Titer korreliert insgesamt mit dem klinischen Bild und kann zum Monitoring der Krankheitsaktivität herangezogen werden (7,19). In einer Studie von Colliou et al. wurde jedoch gezeigt, dass auch Patienten in Vollremission noch erhöhte anti-Dsg3-Titer aufweisen können (12). Obwohl die zentrale Bedeutung von anti- Dsg3-AAK für die Pathogenese von PV von den meisten Autoren akzeptiert wird, scheinen auch Antikörper anderer Spezifität an der Pathogenese von PV beteiligt zu sein. Nguyen et al. konnten nachweisen, dass PV-IgG, die mit dem desmosomalen Strukturprotein Pemphaxin präadsorbiert wurden, keine akantholytische Aktivität im passiven Mausmodel mehr zeigten. Allerdings waren anti-Pemphaxin Antikörper - im Gegensatz zu anti-Dsg3-AAK in anderen Studien - allein nicht ausreichend, um eine Blasenbildung auszulösen (30), sodass ihre pathogenetische Bedeutung unklar bleibt. Um eine mögliche kollaborative Wirkung von anti-Dsg3-AAK und anti-Pemphaxin- AKK zu untersuchen werden weitere Studien benötigt. Zusammenfassend scheinen die an der Entstehung von PV beteiligten AAK gegen verschiedene Antigene gerichtet zu sein, wobei die meisten Studien die besondere pathogenetische Bedeutung der anti- Dsg3-AAK darstellen. Anti-Dsg3-AAK sind in unterschiedlichem Ausmaß pathogen, was bedeutet, dass nicht alle Antikörper eine suprabasale Spaltbildung auslösen. So zeigten Yamagami et al., dass einige anti-Dsg3-AAK, welche aus Patientenseren isoliert wurden, in-vitro keine Blasenbildung auslösten (31). In einer anderen Studie hingegen konnten miteinander kombinierte, ansonsten nicht oder nur niedrig pathogene AAK im Mausmodell und in-vitro eine akantholytische Aktivität entfalten (32). Dieser Ansatz, dass PV auf polyklonale AAK zurückzuführen ist, wird von einer weiteren Studie gestützt, die zeigte, dass monoklonale AK23 im Gegensatz zu polyklonalen PV IgG nicht zu einer Dsg3-Internalisierung führten (33). 5
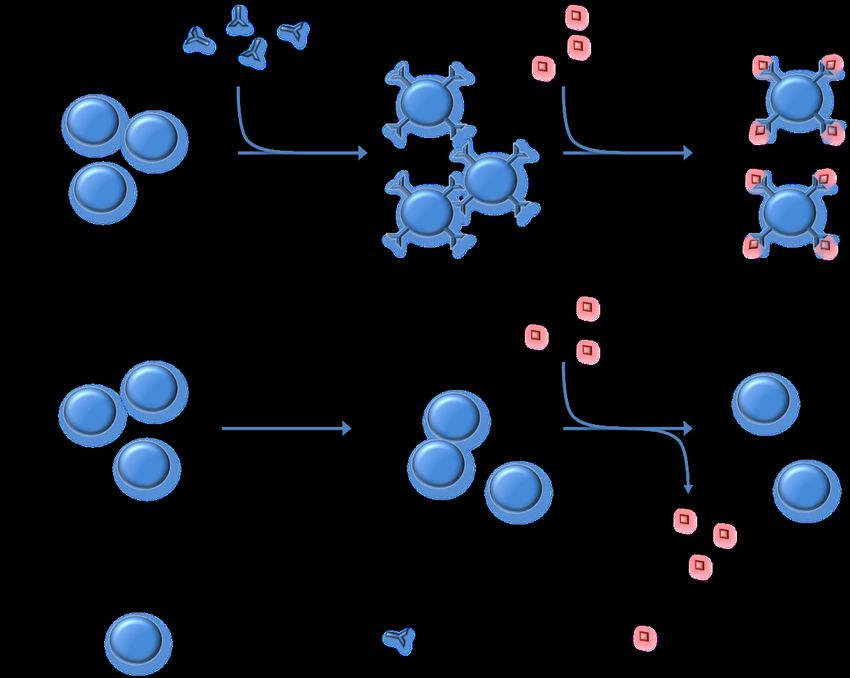
1.3. Selbsttoleranz und ihre Schwachstellen in der B-Zell-Entwicklung Im Rahmen der B-Zell-Entwicklung werden kontinuierlich B-Zellen (BZ) produziert, welche an Autoantigene binden (34,35). Die Anzahl solcher autoreaktiver BZ wird durch Regulationsmechanismen an unterschiedlichen Stationen der BZ-Reifung normalerweise derart begrenzt, dass keine klinisch fassbare Autoimmenreaktion zustande kommt. Es wird dabei konzeptuell zwischen der zentralen Toleranz, die in den frühen Stadien der B-Zell-Entwicklung im Knochenmark (KM) zustande kommt und der peripheren Toleranz, welche in den späteren Entwicklungsschritten in Milz und Lymphknoten induziert wird, unterschieden. Die Mechanismen der Toleranzinduktion beschränken jedoch das mögliche Spektrum der B-Zell-Rezeptoren (BZR), also das antiinfetkiöse Repertoire des Immunsystems. Als Ausdruck eines Kompromisses zwischen Toleranz und Diversität werden nicht alle autoreaktiven BZ eliminiert (34,36,37). Die frühe BZ-Reifung findet im KM statt. Der BZR entsteht hier durch die somatische Rekombination, zuerst der Gene der schweren (VH, DH, JH) und danach der Gene der leichten Kette des BZR (VL, JL ; V(D)J-Rearrangement). Ist die Rekombination erfolgreich, trägt die Zelle BZR des IgM-Typs auf ihrer Oberfläche, wobei die spätere Antikörperklasse (IgM, IgG, IgA, IgD und IgE) durch die Verwendung des jeweiligen Gens für die konstante Region des Antikörpers (CH, CL) bestimmt wird (Abb. 2). Die ersten BZ, welche einen kompletten IgM-Rezeptor tragen, werden naive BZ genannt und entwickeln sich nur zu etwa 30% weiter (38). Ausschlaggebend für die Spezifität des Antikörpermoleküls ist die variable Region mit den drei complementary determining regions 1-3 (CDR1-3), wobei die größte Bedeutung der CDR3 mit ihrer besonders hohen Variabilität zukommt (39). Im Rahmen der zentralen Toleranzinduktion im KM werden autoreaktive BZ von ihrer Umwelt dahingehend beeinflusst, dass sie entweder apoptotisch (sog. klonale Deletion) (40) oder anergisch werden, was bedeutet, dass sie kurzlebiger sind und von 6
T-Helferzellen (TH-Zellen) und ihrem Antigen nur schwer aktiviert werden können (41). Es ist umstritten, ob BZ mit schwacher Bindung an ein Autoantigen, durch welche sie anergisch werden (42), aufgrund möglicher Reaktivierbarkeit ein autoimmunogenes Potenzial bergen (43–46). Trotz der Mechanismen der zentralen Toleranzentwicklung ist ein großer Anteil der frühen BZ im KM noch autoreaktiv (34,35,47,48). Autoreaktive BZ können die somatische Rekombination der leichten und schweren Kette wiederholen um der Apoptose zu entgehen. Dieser, receptor-editing genannte Vorgang, kann sowohl Toleranz induzieren (49–53) als auch Toleranz durch Entstehung autoreaktiver BZR durchbrechen (54) und die Diversität des BZR- Repertoire vergrößern (55): Auf der einen Seite konnten bei Mausmodellen für Systemischen Lupus erythematodes (SLE) und Diabetes mellitus Typ 1 sowie bei Patienten mit diesen Krankheiten eine reduzierte Rate an receptor editing festgestellt werden (56). In anderen Fällen kann receptor editing die Entstehung von Autoimmunität möglicherweise begünstigen: Durch diesen Vorgang entstehen tendenziell längere und damit weniger spezifische CDR3-Regionen (54). Das receptor editing kann zudem zu einer produktiven Rekombination des zweiten Allels und damit zu BZ mit zwei verschiedenen BZR führen (sog. allelische Inklusion) (57). Bei den Autoimmunkrankheiten SLE (58) und Rheumatoider Arthritis (RA) (59) konnte gezeigt werden, dass das Versagen der zentralen Toleranzentwicklung zur Krankheitsentstehung beiträgt. Mit einem funktionstüchtigen IgM-BZR ausgestattete BZ, welche die Kontrollen der zentralen Toleranz passiert haben, verlassen als unreife BZ das KM. In der Peripherie außerhalb der lymphatischen Organe (60) sowie in den sekundären lymphatischen Organen (61) wird die Periphere Toleranz induziert. Deren Induktionsmechanismen unterscheiden sich von denen der zentralen Toleranzentwicklung: Während im KM das receptor editing viele autoreaktive Zellen vor der Apoptose bewahrt, scheint es in den sekundären lymphatischen Organen nur eine untergeordnete Rolle innerhalb der Toleranzentwicklung zu spielen (48,60,62,63), wobei es auch hier stattfindet (64–66). 7
Nach Kontakt mit einem passenden Antigen differenzieren die BZ zu Plasma- oder Gedächtnis-BZ, ändern ihre Antikörper-Klasse (sog. class-switch) und die Affinität ihres BZR reift im Zuge der somatischen Hypermutation (SHM; Abb. 3). Einige Studien zeigen, dass durch den Einfluss der SHM eine Autoantigen-Affinität zunehmen (46,67–69) oder de novo entstehen kann (70,71). Eine Studie von Di Zenzo et al. zeigte, dass auch die Pathogenität von anti-Dsg3-AAK von einer Affinitätsreifung der VH-Domäne durch SHM abhängt: deren Reversion (sog. germlining) hob die Affinität zu Dsg-3 auf (25). Insgesamt gibt es im Laufe der B-Zell-Entwicklung verschiedene Punkte, an denen Autoreaktivität entstehen kann. Möglicherweise ist das Immunsystem von PV Patienten bereits vor Ausbruch der Erkrankung durch Varianten der Keimbahn-DNA für eine solche Entwicklung vulnerabler (72,73). Trotz intensiver Forschung ist jedoch noch nicht geklärt, welche Veränderungen zur Ausbildung pathogener anti-Dsg3- AAK bei Patienten mit PV führen. 8
Abbildung 2: Schematische Darstellung der somatisch-rekombinatorischen Produktion einer schweren Kette. In mehreren Schritten lagert sich je ein Gen jeder Familie (VH, DH, JH, CH) sukzessiv an ein anderes Gen an. Zuerst erfolgt die Anlagerung des DH- an das JH- (a), nachfolgend die des VH- (b) und anschließend die des CH-Gens (d). Diese Gene werden als variable Region (VH; V-, D- und J-Gene) und konstante Region (CH 1-3: CH- Gen) exprimiert (e). Adaptiert und modifiziert aus „Janeway's immunobiology“ (74). 9
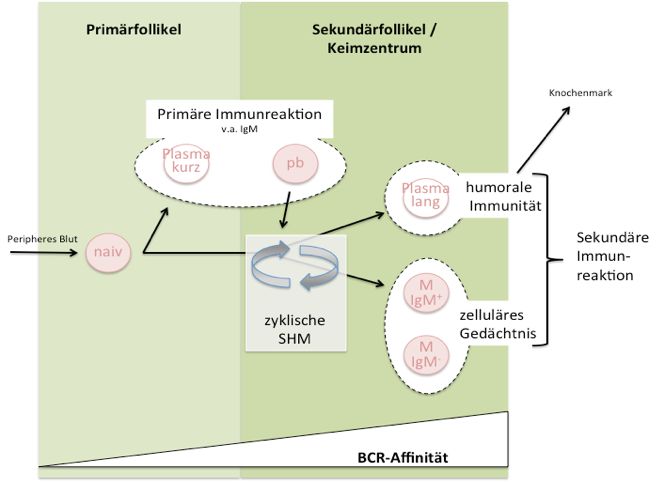
BZR-Affinität Abbildung 3: Entwicklung ausgewählter B-Zell Populationen im Zusammenhang einer Immunreaktion in den sekundären lymphatischen Organen. Eine Antigen- und TH-Zell- aktivierte, naive B Zelle proliferiert klonal, bildet einen Primärfollikel aus und leitet hierdurch eine schnelle Immunreaktion bestehend aus kurzlebigen Plasmazellen (Plasma kurz) und Plasmablasten (pb) ein. Bei adäquater oder wiederholter Antigenexposition können einige der Plasmablasten einen Sekundärfollikel bilden, in dem durch SHM und Selektion hochaffine BZ entstehen können, die entweder zu B-Gedächtniszellen (IgM+, oder andere Ig-Klassen: IgM-) oder langlebige Plasmazellen (Plasma lang) ausdifferenzieren. Die rot ausgefüllten Subpopulationen wurden in den im Folgenden beschriebenen Experimenten untersucht. Sie stellen Entwicklungsstadien dar, die sowohl funktionell als auch im Hinblick auf die Dynamik einer Immunreaktion unterschiedlich sind und einen globalen Eindruck der B-Zell-Population geben sollen. 10
1.4. Genetische Charakteristika der Anti-Dsg3-Immunglobuline Die Untersuchung der Anti-Dsg3-AAK spielt für das Verständnis der Pathogenese von PV eine wichtige Rolle. Diese wurden in verschiedenen Studien anhand ihrer Spezifität isoliert: IgG1 und IgG4 wurden studienübergreifend als die am häufigsten verwendeten Subklassen bei aktivem PV identifiziert (25,75,76). Patienten in Remission hatten ein anderes Verteilungsmuster, je nach Studie wurden vor allem IgG1, IgG2 oder IgG4 gefunden (75,76). In Studien bei denen die phage display Technik angewandt wurde, fand man heraus, dass die Gene der VH-Familien 1, 3 und 4 besonders häufig benutzt werden (25,31,67– 69). In einer Studie von Cho et al. wurden darüber hinaus bei jedem der vier untersuchten Patienten pathologische Dsg3-AAK gefunden, für die das Gen VH1-46 kodierte, welche darüber hinaus wenige bis keine SHM benötigten, um Dsg3 zu binden und pathologische Effekte zu erzielen (68). Das letztgenannte Ergebnis widerspricht jedoch den Aussagen von Di Zenzo et al., welche in ihrer Studie die SHM als Voraussetzung für eine Pathogenität der Antikörper ansahen (25). In einer anderen Studie teilten sich die meisten pathogenen Antikörper ein Aminosäurenmotiv innerhalb der CDR3 Region, welches ebenfalls bei AK23 (24) und nur bei pathogenen AK vorkam (31). Auch bei anderen Autoimmunerkrankungen und interessanterweise auch bei manchen Lymphomen scheint eine VH-Genrestriktion vorzuliegen (77,78). Ein interessanter Aspekt der PV-Pathogenese ist auch, ob Dsg3-spezifische BZ aufgrund ihrer Spezifität selektiert, und einer Affinitätsreifung im Sinne zyklischer SHM unterzogen wurden. Verschiedene Autoren verwenden ein Konzept, bei dem davon ausgegangen wird, dass in den CDR Mutationen zufällig stattfinden. Dabei wird zwischen Mutationen, die eine Aminosäure (AS) ersetzen (Replacement- bzw. R- Mutationen) und solchen, die aufgrund der Redundanz des genetischen Codes keine Änderung der AS-Sequenz nach sich ziehen unterschieden (Silent- bzw. S- Mutationen). Die Sequenz der variablen Domäne wird in mehreren Zyklen immer weiter durch die somatische Hypermutation verändert. R-Mutationen haben einen 11
direkten Einfluss auf die Struktur des BZR. S-Mutationen haben dies nicht, zeigen jedoch an, wie oft die DNA-Sequenz der variablen Domäne einen Zyklus der SHM durchlaufen hat. BZ mit verbesserten Eigenschaften der BZR werden einem positiven Selektionsdruck unterworfen. Insofern zeigen R-Mutationen an, dass Eigenschaften des BZR wie zum Beispiel die Affinität im Vergleich zur Keimbahnsequenz verbessert wurden. S-Mutationen geben Rückschluss darauf, dass das Verbesserungspotential ausgeschöpft ist, was einem negativen Selektionsdruck entspricht (79–81). Die meisten Studien zum Aufbau Dsg3 reaktiver AAK liefern Hinweise für eine solche positive Selektion aufgrund von SHM bei PV (25,67–69). Vor allem der für die Affinität wichtige Abschnitt der VH-Region ist erwartungsgemäß stark mutiert (67). Um zu prüfen, ob die Affinität der Autoantikörper gegen Dsg3 durch SHM erst entstanden ist oder bereits im naiven Zustand vorhanden war, haben Di Zenzo et al. Antikörpersequenzen von immortalisierten, humanen BZ durch sogenannte germline reversion in ihren unmutierten Zustand zurückversetzt. Hierfür wurden die jeweils wahrscheinlichsten Keimbahngene ermittelt, mit ihrer unmutierten Sequenz in-silico zusammengefügt und dann nach Gensynthese rekombinant exprimiert. Dabei zeigte sich, dass die Keimbahnvarianten der VH-Ketten Dsg3 nicht mehr binden konnten, während die germline reversion der VL-Ketten keine Auswirkungen hatte. Dies spricht dafür, dass die Autoreaktivität gegen Dsg3 erst durch SHM entsteht und auf einen Defekt der peripheren Toleranz hinweist (25). 1.5. Desmoglein-3 spezifische B-Zellen Für eine gezielte Therapie des PV ist eine möglichst genaue Charakterisierung der an der Pathogenese beteiligten BZ von großem Interesse. Aufgrund der wichtigen Rolle der Dsg3-spezifischen AAK widmen sich viele Studien der Klonalität, der Antikörperklasse und dem Einfluss der SHM bei diesen AAK, da von diesen Parametern auf eine Reihe von Merkmalen der AAK sezernierenden BZ geschlossen 12
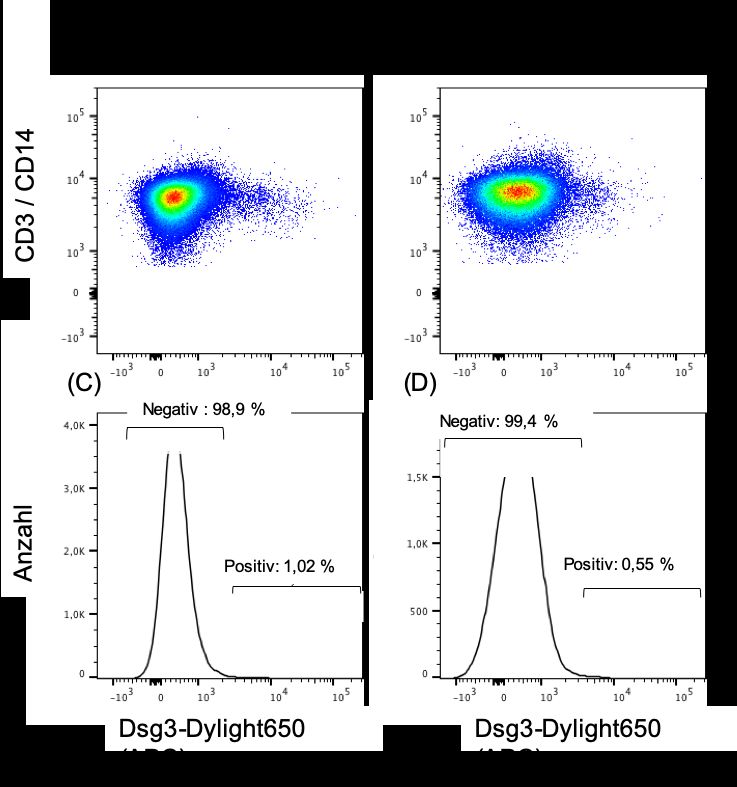
werden kann: Studien an anti-Dsg3-AAK aus dem peripherem Blut von PV Patienten zeigten, dass diese stark hypermutiert (25,69) und vom IgG-Subtyp sind (75,76), sodass geschlossen werden kann, dass die sezernierenden BZ einen Klassenwechsel und SHM-Zyklen durchlaufen haben. In mehreren Studien wurde zudem versucht, die AAK produzierenden, Dsg3 spezifischen BZ direkt zu detektieren: Bei dem Ansatz, den Colliou et al. und Yamagami et al. verfolgten, wurden PBMC von PV Patienten mit markiertem Dsg3 inkubiert. BZ, die Dsg3 gebunden hatten, wurden in einem zweiten Schritt durchflusszytometrisch quantifiziert und weiter charakterisiert (13). Ye Qian et al. hingegen immortalisierten PBMC von PV Patienten, indem sie diese mit Myelom- Zellen fusionierten. Die daraus etablierten Zellkulturen wurden im Verlauf auf das Vorhandensein von anti-Dsg3-AK überprüft und positive Kulturen konsekutiv weiter vereinzelt. Die so isolierten anti-Dsg3-AK produzierenden BZ konnten danach weiter charakterisiert werden (67). Auf dem Boden dieser Untersuchungen zeigte sich, dass diese Zellen im Blut von PV Patienten mit einer Frequenz von 1-18 pro 105 PBMC auftreten (82) und, im Gegensatz zu den allermeisten anti-Dsg3-AK, IgM-positiv sind (13,67). In der oben genannten Studie von Colliou et al. (13) wurde die Dynamik von Dsg3- AAK und Dsg3-spezifischen BZ unter Therapie mit dem BZ depletierenden CD20- Antikörper Rituximab untersucht: Nach initialer BZ-Depletion zeigten Patienten mit kompletter Remission (CR) im Gegensatz zu Patienten mit inkompletter Remission (IR) nach 6 Jahren eine sehr niedrige Anzahl Dsg3-spezifischer IgG+ BZ und kaum nachweisbare anti-Dsg3-AAK Titer im peripheren Blut. Die Anzahl Dsg3-spezifischer IgM+ BZ lag in beiden Kohorten nach 6 Jahren auf (CR) oder über dem Ausgangswert (IR). Daraus lässt sich schließen, dass IgG+ BZ für den Krankheitsverlauf eine größere Bedeutung haben als IgM+ BZ. Eine mögliche Erklärung für das heterogene klinische Ansprechen könnte sein, dass bei den Patienten mit IR ein Übergang der AAK produzierenden BZ Population zu sogenannten langlebigen PC (83) stattgefunden haben könnte, welche aufgrund eines fehlenden CD20-Rezeptors durch die 13
Rituximab-Therapie nicht erfasst wurden. Für diesen Zusammenhang sprechen eine Reihe weiterer Beobachtungen: Zum einen waren die anti-infektiösen IgG (84), welche von langlebigen PC sezerniert werden (85), bei allen Patienten konstant (13,86,87), was bedeutet, dass langlebige Plasmazellen nicht depletiert wurden. Zum anderen deutet das gute Ansprechen bei früher Rituximab-Gabe (86) oder als Ersttherapeutikum (13) in anderen Therapiestudien darauf hin, dass zu einem frühen Zeitpunkt viele CD20+ autoreaktive BZ eliminiert werden können. Ob Patienten mit längerem Krankheitsverlauf auf Rituximab aufgrund einer langlebigen CD20- BZ-Population schlechter ansprechen bleibt hingegen offen. Eine Langzeitremission durch die Depletion pathogener BZ ist vermutlich vereinzelt möglich, da nicht ständig neue Dsg3-spezifische BZ produziert werden, sondern pathogene BZ-Klone über Jahre persistieren und die Patienten nach Depletion dieser Zellen in Remission gehen (88). Diese Oligoklonalität Antikörper-sezernierender und zirkulierender Dsg3-spezifischer BZ wurde bereits in verschiedenen Studien gezeigt (67,82,89). Bisher unklar ist, welche Rolle IgM+ BZ bei der Pathogenese und im Krankheitsverlauf von PV spielen. 1.6. BZR-Repertoires Der Begriff BZR-Repertoire beschreibt die Gesamtheit aller BZR, die sich in den für die Immunglobuline kodierenden genetischen Sequenzen wiederspiegelt. Dabei ist zu beachten, dass aufgrund der Redundanz des genetischen Codes mehrere genetische Sequenzen für dieselbe Aminosäurekette kodieren können. Das BZR-Repertoire rekrutiert sich aus BZ-Populationen, die eine variable Ähnlichkeit zueinander haben, je nachdem ob sie sich eine Keimbahnsequenz (germline sequence) oder darüber hinaus auch somatische Mutationen teilen. Solche Gruppen nennt man auch Klonotypen. Anhand der Anzahl solcher geteilter Merkmale kann man das Repertoire nach der Abstammung der Sequenzen ordnen und Stammbäume erstellen. 14
Das BZR-Repertoire einer Probe kann anhand der Sequenzierung der RNA oder DNA von BZ erfasst werden. Es besteht dabei die Möglichkeit die schwere oder die leichte Kette, bzw. nur die CDR3 der schweren Kette zu sequenzieren. Die letztgenannte Möglichkeit ermöglicht trotz eines geringeren technischen Aufwandes eine Diskriminierung unterschiedlicher BZ-Klonotypen und der an der Antigenbindung beteiligten AS sowie das Beurteilen des Einflusses der SHM (90). Dies ist möglich, da die Spezifität des Ig maßgeblich von der CDR3H aufgrund ihrer sehr hohen Variabilität abhängt (39). Neben der SHM kommt die Variabilität durch insgesamt 51 VH-, 27 DH- und 6 JH-Gene zustande, bei deren Kombination verschiedene Übergangssequenzen entstehen und noch zufällig Nukleotide addiert werden. Im Gegensatz dazu werden die CDR1H und CDR2H ausschließlich vom jeweiligen VH-Gen kodiert. Ein Nachteil der alleinigen CDR3H-Sequenzierung ist die fehlende Möglichkeit, anhand der Sequenz die Ig-Klasse zu bestimmen. Dieser Nachteil lässt sich jedoch durch vorheriges Sortieren der Zellen ausgleichen. Ebenfalls ist die Methode zur Auswahl der Zellen maßgeblich am Ergebnis beteiligt. Das sogenannte bulk sequencing beschreibt die Analyse der DNA einer ganzen, z.B. durchflusszytometrisch isolierten Zellpopulation (91). Ein Vorteil dieser Methode besteht in der hohen Ausbeute an Sequenzen, ein Nachteil ist die fehlende Möglichkeit, bei Sequenzierung beider Ketten deren jeweilige Paarung zu ermitteln. Die Sequenzierung der Ig-DNA einzelner Zellen ermöglicht die Bestimmung der IgH/IgL-Paarung. Aufgrund des hohen Aufwands können jedoch keine vergleichbar großen Populationen untersucht werden (92). Dieser Nachteil gilt auch für das phage display, eine Methode, bei der BZR-Sequenzen aus isolierten BZ in Bakteriophagen eingebracht und anschließend in mehreren Runden anhand ihrer AG-Spezifität selektiert werden (93). Bei dieser Technik ist ebenfalls keine Bestimmung der ursprünglichen IgH/IgL-Paarung möglich (94) . 15
1.7. Fragestellung Pemphigus vulgaris (PV) ist eine Blasen-bildende Autoimmunerkrankung der Haut, die durch Autoantikörper produzierende, Desmoglein-3-spezifische B-Zellen vermittelt wird. Bisherige Studien kamen zu heterogenen und teilweise widersprüchlichen Ergebnissen hinsichtlich genetischer Charakteristika der Desmoglein-3-spezifischen Autoantikörper und der zirkulierenden Desmoglein-3- spezifischen B-Zellen (13,25,67,75,76). Trotz der vermutlich zentralen Bedeutung dieser Zellen für die Pathogenese von PV erfolgte ferner bislang keine Zuordnung zu einer definierten B-Zell-Subpopulation. Die in dieser Arbeit untersuchte Hypothese war, dass bei der systemischen Autoimmunerkrankung PV eine Dysregulation des globalen B-Zell- Rezeptorrepertoires der Patienten vorliegt. Mithilfe einer zu entwickelnden durchflusszytometrischen Sortierungsmethode, die eine Auftrennung der B-Zellen anhand ihres Immunphänotyps in vier definierte Subpopulationen verschiedener Entwicklungsstufen ermöglicht, sollten B-Zellen aus dem peripheren Blut von 10 PV- Patienten und von 10 gesunden Kontrollen analysiert und separiert werden. Die genomische DNA dieser Subpopulationen sollte im Anschluss extrahiert und die B- Zell-Rezeptorgene in einem Hochdurchsatzverfahren (next generation sequencing; NGS) sequenziert werden, um die B-Zell-Rezeptorrepertoires der Studiengruppen mit Hilfe bioinformatischer Analysen zu vergleichen und PV-spezifische Veränderungen zu identifizieren. Das zweite Ziel dieser Arbeit war die Detektion, Quantifizierung und Immunphänotypisierung Desmoglein-3-spezifischer B-Zellen im Blut von PV- Patienten. Zu diesem Zweck sollte das Antikörper-Panel des zuvor etablierten durchflusszytometrischen Verfahren um fluoreszenzmarkiertes Desmoglein-3 erweitert werden. 16
2. Material und Methoden 2.1. Material 2.1.1. Geräte und Laborbedarf Name Hersteller Pipetten Research®, Eppendorf, Hamburg, Deutschland 0,5-10 µl, 2-20 µl, 10-100 µl, 100-1000 µl Multipipetten Eppendorf, Hamburg, Deutschland 0,5-10 µl, 10-100 µl, 30-300 µl pipetus-Pipettierhilfe Hirschmann, Eberstadt, Dutschland Pipettenspitzen Sarstedt, Nümbrecht, Dutschland 10 µl, 100 µl, 1000 µl ELISA-Tips Eppendorf, Hamburg, Deutschland 2-200 µl, 20-300 µl Safe-Seal-Gefäße Sarstedt, Nümbrecht, Deutschland 1,5 ml, 2 ml Serologische Pipetten Sarstedt, Nümbrecht, Deutschland 5 ml,10 ml, 25 ml FACS-Röhrchen Sarstedt, Nümbrecht, Deutschland Pasteur-Glaspipette Sarstedt, Nümbrecht, Deutschland Filtopur V50 Filter Sarstedt, Nümbrecht, Deutschland MACS-Filter Miltenyi Biotec, Bergisch Gladbach, Deutschland nunc-Immuno-Plate Maxisorp-Surface Nalge-nunc international, Rochester, 96 well flat bottom USA Klebefolie, optisch-klar Sarstedt, Nümbrecht, Deutschland S-Monovette EDTA 9 ml Sarstedt, Nümbrecht, Deutschland Safety-Multifly-Kanüle Sarstedt, Nümbrecht, Deutschland Cutasept F Hautdesinfizienz Bode, Hamburg, Deutschland Megafuge 1.0R Heraeus instruments, Hanau, Deutschland Centrifuge 5810 R Eppendorf, Hamburg, Deutschland Biofuge fresco Heraeus instruments, Hanau, Deutschland Bio-Vortex V1 BOECO, Hamburg, Deutschland Vortex-Genie 2 Scientific Industries, Bohemia, USA Reac 2000 Vortex Heidolph, Schwabach, Deutschland 17
Testplattenmischer TPM 4 Sarstedt, Nümbrecht, Deutschland Rührfeld CB162 Stuart, Stone, Vereinigtes Königreich pH-Meter pH562 WTW, Weilheim, Deutschland Wasserbad Köttermann, Uetze, Deutschland Wilovert S Mikroskop Hund Wetzlar, Wetzlar, Deutschland LaminAir HB 2448 sterile Abzugshaube Heraeus instruments, Hanau, Deutschland Biowizard sterile Abzugshaube Kojair, Vilppula, Finnland captair chem Abzugshaube Erlab, Paris, Frankreich Analytical Plus Feinwaage OHAUS, Parsippani, USA EMB 220-1 Waage Kern & Sohn, Balingen, Deutschland Neubauer-Zählkammer Marienfeld, Lauda Königshofen, Deutschland Victor 3 PerkinElmer, Waltham, USA FACS Aria III Durchflusszytometer, BD biosciences, San Jose, USA Laser: 561 nm, 488 nm, 633 nm 405 nm Qubit Fluorometer Thermo Fisher Scientific, Grand Island, USA Illumina MiSeq Illumina, San Diego, USA Bioanalyzer 2100 Agilent Technologies, Santa Clara, USA 18
2.1.2. Puffer, Medien und Lösungen Name Hersteller Zusammensetzung PBS (Phosphate-buffered Eigenherstellung 140 mM NaCl, Saline) 10,0 mM Na2HPO4, 2,7 mM KCl 1,8 mM KH2PO4 ad pH 7,3 mit H3PO4 Eigenherstellung 50 mM Tris, 0,14 M NaCl, TBS (Tris-buffered Saline) 1 % BSA, 0.05 % Tween, + 0,4 mM Ca2+ auf pH 8.0 eingestellt mit HCl Lysepuffer Eigenherstellung Ampuwa, 0,9 % NaCl (4:1) Lymphoprep™ STEMCELL, Vancouver, Kanada RPMI-1640 Thermo Fisher Scientific, Waltham, USA DPBS (Dulbecco's Thermo Fisher Scientific, Phosphate-Buffered Waltham, USA Saline) Beschichtungspuffer Eigenherstellung 0.05 M Natriumkarbonat, 0,05 M Natriumbikarbonat, auf pH 9.6 eingestellt mit HCl DNA stabilisierende Zell- Qiagen, Hilden, Lyse-Puffer Deutschland 19
2.1.3. Chemikalien Alle Chemikalien wurden in der Qualität pro analysi verwendet. Name Hersteller Ethanol 70 % vergällt Carl Roth, Karlsruhe, Deutschland Aqua ad iniectabilia, Ampuwa Fresenius Kabi, Bad Homburg, Deutschland Natriumcarbonat, wasserfrei Merck, Darmstadt, Deutschland Na2CO3 Natriumhydrogencarbonat (NaHCO3) Merck, Darmstadt, Deutschland TRIS (Hydromethylaminomethan, Serva, Heidelberg, Deutschland C4H11NO3) Tween 20 (C58H114O26) Merck, Darmstadt, Deutschland Natriumchlorid (NaCl) Carl Roth, Karlsruhe, Deutschland Salzsäure 2M (HCl) Carl Roth, Karlsruhe, Deutschland Natronlauge 2M (NaOH) Merck, Darmstadt, Deutschland Turbo-TMB-ELISA Schwefelsäure 25 % Merck, Darmstadt, Deutschland (H2SO4) Turbo-TMB-ELISA Thermo Fisher Scientific, Waltham, USA IPTG (Isopropyl-β-D- Thermo Fisher Scientific, Waltham, thiogalactopyranosid, C9H18O5S) USA Albumin Fraktion V, biotinfrei Carl Roth, Karlsruhe, Deutschland 2.1.4. Kits und Enzyme Name Hersteller Gentra PureGene Blut Kit Qiagen, Hilden, Deutschland AmpliTaq Gold Life Technologies, Carlsbad, USA Agencourt AMPure XP bead system Beckman Coulter, Inc., Indianapolis, USA MiSeq Reagenzien Kit v3 Illumina, San Diego, USA 20
2.1.5. Antikörper, Konjugatfarbstoffe, Kompensationshilfen Name Hersteller Murin anti-human Desmoglein-3 (Klon: Eigenherstellung AK23) Murin anti-human Dsg3 (Isotyp IgG1, Thermo Fisher Scientific, Waltham, Klon: 5G11), c = 0,5 mg / ml USA Murin Anti-penta-His (Isotyp IgG1, Thermo Fisher Scientific, Waltham, Klon: USP7), c = 0,2 mg / ml USA FITC Ratte-anti-Maus-IgG (IgG Thermo Fisher Scientific, Waltham, polyklonal, Katalog Nr.: 11-4011-85), USA c = 0,5 mg / ml Ziege FITC anti-human-IgG (IgG Thermo Fisher Scientific, Waltham, polyklonal, Katalog Nr.: 62-8411), USA c = 0,6 mg / ml Murin APC anti-human CD19 (IgG1 BD biosciences, San Jose, USA kappa, Klon: HIB19), c = 5 Tests / µl Biolegend (IgG1 kappa, Klon: MHM88), BioLegend, San Diego, USA c = 0,5 mg / ml Brilliant Violet 605TM Murin anti- BioLegend, San Diego, USA human CD27 (IgG1 kappa, Klon: O323), c = 10 Tests / µl PE/Cy7 Murin anti-human CD19 (IgG1 BioLegend, San Diego, USA kappa, Klon: IHB19), c = 0,2 Tests / µl PE Murin anti-human CD38 (IgG1 BioLegend, San Diego, USA kappa, Klon: Hb7), c = 0,2 Tests / µl FITC Murin anti-human CD3 (IgG2a BioLegend, San Diego, USA kappa, Klon: HIT3a), c = 0,2 Tests / µl FITC Murin anti-human CD14 (IgG2a BioLegend, San Diego, USA kappa, Klon: M5E2), c = 0,2 Tests / µl DyLight™ 650 NHS Ester Thermo Fisher Scientific, Waltham, USA BD™ CompBeads Set Anti-Mouse Ig, κ BD biosciences, San Jose, USA Desmoglein-3, extrazelluläre Domains, EuroImmun, Lübeck, Deutschland His-tagged 21
2.1.6. Ethischer und datenschutzrechtlicher Umgang mit Patientenmaterialien In der vorliegenden Dissertation wurden Blut- und DNA-Proben von Patienten mit PV sowie von gesunden Spendern verwendet und gelagert. Für diese Studie wurde die Billigung durch die Ethikkommission der Universität zu Lübeck unter dem Aktenzeichen Nr 07-179 eingeholt. Die Studie wurde gemäß der Deklaration von Helsinki und geltender Datenschutzbestimmungen durchgeführt. Alle PV-Patienten und gesunden Spender wurden sorgfältig über die Studie und die Verwendung ihrer Materialien aufgeklärt und gaben ihr schriftliches Einverständnis. 22
2.2. Methoden 2.2.1. Herstellung von Desmoglein-3-Dylight650 Zur Herstellung eines Konjugats aus Dsg3 und dem Fluoreszenzfarbstoff Dylight650 wurde 50 µg Dylight650 in Pulverform in 500 µl einer Lösung aus 0,7 mg / ml Dsg3 in Tris-buffered-saline (TBS) aufgelöst. Danach wurde die Lösung für 5 sec gemischt und für eine Stunde lichtgeschützt bei Raumtemperatur (RT) inkubiert. Anschließend wurden ungebundene Dylight650-Moleküle durch dreimalige Dialysierung mit TBS als Dialysemedium ausgewaschen. Im Anschluss wurde eine 10 : 1 Verdünnung der Antigenlösung mit TBS erstellt und diese in einer Küvette bei 655 nm und 288 nm photometrisch untersucht. Anhand des vom Hersteller bereitgestellten Schemas wurde anschließend die durchschnittliche Frequenz der Farbstoffmoleküle pro Antigenmolekül ermittelt. 2.2.2. Desmoglein-3-spezifischer ELISA Der Dsg3-spezifische Enzyme-linked Immunosorbent Assay (ELISA) wurde mit 72-Loch- Platten durchgeführt. Diese wurden mit 50 µl Beschichtungspuffer mit einer Konzentration von 5 µg / ml Antigen, jeweils gefärbt und ungefärbt, befüllt. Es folgte die Inkubation über Nacht bei 4 °C. Danach wurde die Platte fünf Mal mit 300 µl TBS gewaschen. Anschließend wurden Primärantikörper in 200 µl TBS gelöst aufgetragen. Als Testseren wurden die aufgereinigten IgG zweier Personen mit aktivem PV benutzt (jeweils in den Verdünnungen 1 : 51, 1 : 510 und 1 : 5100). Als Positivkontrollen wurden murine anti-Dsg3-AK (AK23, 1 : 200 verdünnt) und murine anti-penta-his-AK (1 : 200 verdünnt, Bindung an das His-Tag des Dsg3) aufgetragen. TBS alleine oder IgG eines gesunden Spenders (Verdünnungen 1 : 51, 1 : 510 und 1 : 5100) dienten als Negativkontrolle. Es folgte eine Inkubation (30 Minuten, RT). Nach einem weiteren 23
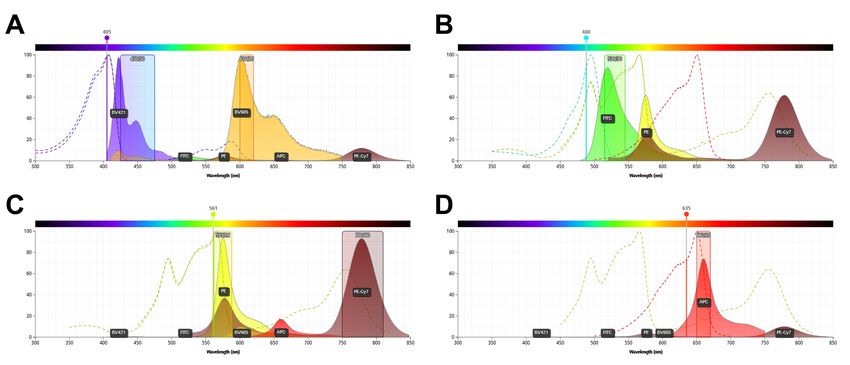
Waschschritt wurden dann 50 µl der gegen Maus- bzw. gegen humanes IgG gerichtete, Horseraddish-peroxidase-markierte AK in einer Verdünnung von 1 : 500 appliziert. Nach einer weiteren Inkubation (30 Minuten, RT) wurden die Platten gewaschen, die Proben durch Zugabe von TMB-Turbo gefärbt und danach mit 100 µl 25 %-iger Schwefelsäure fixiert. 2.2.3. Durchflusszytometrische Evaluation des Desmoglein-3-Dylight650 Um die die Bindung des Dsg3 an Dylight650 zu verifizieren erfolgte eine Messung im Durchflusszytometer mithilfe von CompBeads. Zuerst wurden 250 µl Tris-Puffer in 1,5 ml Eppendorf-Reaktionsgefäßen vorgelegt. Danach wurde je ein Tropfen CompBeads (nicht bindend / bindend) hinzugefügt. Bei CompBeads handelt es sich um Kunststoffkugeln, die murine Antikörper binden, in etwa die Größe menschlicher Zellen haben und somit als Kompensationshilfen für durchflusszytometrische Experimente genutzt werden können. Es wurden 0,5 µl anti- Dsg3-AK (Murin anti-human Dsg3 ; Isotyp IgG1, Klon: 5G11) hinzugefügt, die Proben für 10 min bei RT abgedunkelt inkubiert und danach zentrifugiert (5 min, 2000 rpm, RT). Der Überstand wurde verworfen, die Proben in 100 µl Puffer resuspendiert und vor dem nächsten Schritt noch einmal ebenso gewaschen. Danach wurde entweder gefärbtes Dsg3 oder Sekundärantikörper zur Kontrolle aufgetragen: 4 µl anti-CD19- APC, 1 µl anti-Maus-IgG-FITC und Dsg3-DL650 in den Mengen 0,25 µl, 0,5 µl und 1 µl. Abschließend wurden die Proben in 250 µl Puffer resuspendiert und durchflusszytometrisch ausgewertet. 24
2.2.4. Vorbereitung humanen Vollblutes für die durchflusszytometrische Untersuchung Es wurden sechs 45 ml Falcon Röhrchen mit 15 ml RPMI und weitere sechs 45 ml Falcon Röhrchen mit 10 ml Lymphoprep befüllt und auf Raumtemperatur aufgewärmt. Die zuvor gewonnenen 45 ml Vollblut wurden dann auf die sechs mit RPMI gefüllten Röhrchen verteilt. Nach leichtem Schwenken der Röhrchen wurden die 24 ml des RPMI-Blut-Gemisches langsam auf die vorbereiteten 10 ml Lymphoprep geschichtet. Die Proben wurden zentrifugiert (25 min, 1600 rpm, RT). Danach wurde die Lymphozytenschichten der Proben mit einer 5 ml Pasteurpipette aufgenommen, in einem 45 ml Falcon Röhrchen gesammelt und dieses Röhrchen mit RPMI auf 45 ml aufgefüllt. Alle folgenden Schritte wurden auf Eis, bzw. bei 4 °C durchgeführt: Nach einer weiteren Zentrifugation (15 min, 1600 rpm) wurde der Überstand abgesaugt und verworfen, das Zellsediment mit 25 ml kaltem RPMI Medium resuspendiert und wie im vorherigen Schritt zentrifugiert. Danach wurden verbliebene Erythrozyten im Zellsediment mit 4 ml Lysepuffer für 30 s lysiert, das Röhrchen, um die Lyse zu stoppen auf 25 ml mit HBSS aufgefüllt, und die Probe erneut zentrifugiert. Nach Entfernen des Überstandes wurden die Zellen in 1 ml FACS-Puffer aufgenommen und in einer Neubauer-Zählkammer gezählt. In ein 15 ml Falcon-Röhrchen wurden TBS, Antikörper (Mengenangaben siehe Tab. 1) und Dsg3 pipettiert. Danach wurden 4 x 107 PBMC hinzugefügt, das Gefäß einige Male geschwenkt und für 30 Minuten auf Eis lichtgeschützt inkubiert. Es folgte eine Zentrifugation (1700 rpm, 4 °C, 5 min). Nach Absaugen des Überstandes wurde das Zellsediment in 1 ml TBS resuspendiert und auf einen MACS Presorting Filter gegeben. Der Durchfluss wurde in ein FACS-Röhrchen überführt danach analysiert. Zu Kompensationszwecken wurden für jeden Farbstoff eine Einzelfärbung mit CompBeads erstellt. Dafür wurden 200 µl TBS in je ein 1,5 ml Eppendorf- 25
Reaktionsgefäß gefüllt, je ein Tropfen der Antikörper-bindenden und der nicht- bindenden CompBeads sowie das angegebene Volumen an Antikörper hinzugefügt. Inkubation und Zentrifugation wurden wie oben beschrieben durchgeführt. Für die Analyse wurden die Proben in 300 µl TBS gelöst. Spezifität Farbstoff μl / 10 ^ 6 Zellen CD3 FITC 1 CD14 FITC 1 CD19 PE-Cy7 1 CD27 BV605 1 IgM BV421 0.5 CD38 PE 0.5 Tabelle 1: Antikörper-Panel zur Separation von BZ-Subpopulationen 2.2.5. DNA-Extraktion, Amplifizierung per PCR und Sequenzierung der schweren Kette der B-Zell-Subpopulationen Die sortierten Zellen wurden in DNA stabilisierender Lösung für maximal 6 Monate bei 4 °C gelagert, anschließend wurde die genomische DNA mit dem Gentra PureGene Blood Kit extrahiert (jeweils nach den Empfehlungen des Herstellers). Für die Vervielfältigung der genomischen DNA der VH-Gene per Polymerasekettenreaktion (polymerase chain reaction, PCR) wurden zwei unterschiedliche Primertypen benutzt, welche die V-, D- und J-Genloci der schweren Kette abdeckten (nach Meng et al. und van Dongen et al. (95,96)). Es wurden zwei Ansätze aus vorwärts-gerichteten Primern (forward primer) benutzt, die an unterschiedliche Regionen der einzelnen VH- Subklassen binden. Diese wurden jeweils mit einem rückwärtsgerichteten JH Primer (reverse primer) kombiniert. Die forward Primer des einen Ansatzes banden an die framework-1-Region (FWR1-Primer), die des anderen an die Leader Region der VH Gene (Leader-Primer). Die so eingefassten VDJ-Regionen zwischen den Primern wurden mit der Taq-Polymerase AmpliTaq Gold (Life Technologies, Carlsbad, USA) in einer 26
zehnfach verdünnten PCR-Pufferlösung mit resultierenden Konzentrationen von 1.5 mM MgCl2, 0.2 mM Desoxyribonukleotid-triphosphat (dNTPs) und 0.5 uM Primermix amplifiziert. Das DNA-Produkt wurde danach anhand des Agencourt AMPure XP bead system (Beckman Coulter, Inc., Indianapolis, IN) aufgereinigt, die Qualität der PCR-Bibliothek mit dem Bioanalyzer 2100 (Agilent Technologies, Santa Clara, USA) überprüft und per Qubit Fluorometer (Thermo Fisher Scientific, Grand Island, USA) quantifiziert. Danach wurden die Amplikons mit sogenannten Barcodes versehen und mit einem Illumina MiSeq System und dem MiSeq Reagenzien Kit v3 sequenziert (beides Illumina, San Diego, USA). Das System beruht auf der Sequenzierung-durch-Synthetisierung-Methode (sequencing by synthesis). 2.2.6. Statistische Analyse Die statistischen Analysen wurden mit dem Statistikprogramm „R“ (The R Foundation for Statistical Computing) unter Verwendung verschiedener Software- Pakete durchgeführt. Die genauen statistischen Methoden sind im Ergebnisteil aufgeführt. Bei Analysen, welche Mittelwerte zwischen Patienten und Kontrollen vergleichen, wurden die Ergebnisse nach Geschlecht stratifiziert um eine Fehlwichtung durch die unterschiedliche Geschlechterverteilung in den beiden Gruppen auszugleichen. Als grundsätzliches Testverfahren wurde in dieser Arbeit der Permutationstest verwendet, da hierbei eine Berücksichtigung von relevanten Stratifikationsfaktoren wie dem Geschlecht einfach umsetzbar ist. Permutationsteste sind im R-Paket coin implementiert. Für besondere Fragestellungen wurden eigene Testverfahren in R programmiert, wie beim Cosinusdistanztest. Beim Permutationstest wird der Wert einer Zielstatistik aus dem Vergleich zweier Gruppen verglichen mit den Werten nach einer Zufallsdurchmischung der Gruppen (Resampling). Wenn beispielsweise das Geschlecht als Stratifikationsfaktor berücksichtigt werden sollte, wurde diese Zufallsdurchmischung so vorgenommen, dass nur Männer bzw. Frauen zwischen den 27
Vergleichsgruppen ausgetauscht (permutiert) wurden. Ein p-Wert von < 0,05 wurde erreicht, wenn der Wert der Statistik für die Originaldaten größer bzw. kleiner ist als der von 95% der permutierten Daten. 2.2.7. Definition von Klonen und Bestimmung der Klon-Größe Die Diskriminierung von klonalen DNA-Sequenzen erfolgte nach einem Schema, welches sich an dem von Hershberg und Luning Prak orientiert (94): Die DNA- Sequenzen wurden mit dem Programm pRESTO sortiert, Sequenzen mit einem Qualitätsscore (Phred-score) von >20 wurden weiter verwendet (97). Anschließend wurden komplementäre Vorwärts- und Rückwärtsstränge gepaart und Sequenzen unter 10 Basenpaaren ausgeschlossen. Mittels IMGT/HighV-QUEST-Plattform (The international ImMunoGeneTics information system, High throughput V-QUERy and STandardization, URL: http://www.imgt.org) wurden die VH-, DH- und JH-Gene sowie Frequenz und Art der in diesem Bereich aufgetretenen Mutationen ermittelt (98). Nun wurden die individuellen Sequenzen in Abhängigkeit von ihrer Häufigkeit sortiert. Sequenzen mit gleichem VH- und JH-Gen sowie 85% Übereinstimmung der Basensequenz (relative Hamming-Distanz) wurden dann als Klon definiert und die Klongrößen in einer Untergruppe von 1000 Sequenzen mit der Funktion rarefy des Statistikprogramms „R“ (Paket: vegan) ermittelt. Danach konnten die Klone nach Größe sortiert werden, wobei wir die Sequenzen beider Primerpaare gesondert analysierten. Die Diversität der Sequenzen wurde mit der Funktion diversity ermittelt. 2.2.8. Analyse der Verteilung und Variabilität der VH- und DH-Genbenutzung Um die große Variabilität der VH- und DH-Gene darzustellen, wurde die von-Mises- Fisher-Verteilung benutzt. Dabei werden die Parameter einer Probe als Dimensionen einer n-dimensionalen Sphäre definiert, aus deren unterschiedlichen Werten sich ein Richtungsvektor ergibt. Eine Gruppe von Vektoren kann dann durch den 28
Sie können auch lesen

















































