EVOLUTION IM ZEITRAFFER - SARS-COV-2-VARIANTEN - DEUTSCHES ÄRZTEBLATT
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
MEDIZINREPORT
Thema SARS-CoV-2-Varianten
Evolution im Zeitraffer
Für den Fortgang des pandemischen Geschehens spielen Fragen zu Eigenschaften, Verbreitung und
Bedeutung mutierter Varianten von SARS-CoV-2 eine große Rolle. Die molekulare Surveillance erfolgt
am Robert Koch-Institut unter anderem mittels Gesamtgenomsequenzierungen.
D
as Betacoronavirus SARS- onsartige weltweite Verbreitung. Ba- men sogenannter Superspread-Er-
CoV-2 gelangte höchstwahr- sierend auf den genomischen Unter- eignisse kann es beispielsweise zur
scheinlich als zoonotischer schieden sind weltweit mittlerweile weitreichenden Ausbreitung eines
Erreger gegen Ende 2019 in die mindestens 9 verschiedene Kladen Virustyps kommen, der anschlie-
menschliche Bevölkerung, vermut- bestehend aus 1 021 verschiedenen ßend die Viruspopulation dominiert;
lich in Form einer einzelnen Trans- Viruslinien beziehungsweise -vari- in geografisch abgegrenzten Gebie-
mission (monophyletischer Ur- anten definiert (3). Diese unterschei- ten können „founder effects“ zum
sprung) aus einem noch immer un- den sich in definierten Positionen Tragen kommen, bei denen die re-
bekannten Tierreservoir (1, 2). Eben- der viralen Erbinformation, die mit gionale Viruspopulation von weni-
so wie zum Beispiel bei Influenza- Veränderungen der kodierten Virus- gen „Gründerviren“ abstammt und
viren und HIV handelt es sich bei proteine und damit besonderen Ei- sich in ihrer Zusammensetzung er-
SARS-CoV-2 um ein RNA-Virus, genschaften der jeweiligen Virus- heblich von den Viruspopulationen
dessen Genom durch eine viral ko- linie einhergehen können. in anderen Regionen unterscheiden
dierte RNA-abhängige RNA-Poly- kann (6). Viele der beobachteten
merase repliziert wird. Trotz der Eindeutige Identifikation Mutationen wirken sich nicht er-
Korrekturaktivität der viralen Exo- Die Zuordnung zu bekannten oder kennbar auf die Viruseigenschaften
nuklease nsp14 verläuft die Repli- neuen Viruslinien kann zweifelsfrei aus. Mitunter führen die Mutationen
kation des viralen Genoms nicht nur über eine Sequenzierung des jedoch zu Änderungen in viralen
fehlerfrei. Dies begünstigt Kopier- vollständigen Virusgenoms erfol- Proteinen (z. B. des Spike-Rezep-
fehler und damit den Erwerb von gen. Von den 3 parallel verwende- torbindeproteins), die sich auf die
Mutationen im viralen Genom. ten Nomenklatursystemen hat jedes biologischen Erregereigenschaften
Die Wahrscheinlichkeit des Auf- bestimmte Stärken, zur Feindifferen- (z. B. gesteigerte Infektiosität durch
tretens solcher Mutationen hängt da- zierung wird derzeit sehr häufig die erhöhte Rezeptorbindeaffinität) oder
bei von verschiedenen Faktoren ab, phylogenetische PANGOLIN-Klas- auf die Erkennung durch das
insbesondere der Fehlerrate der vira- sifizierung nach Rambaut verwen- menschliche Immunsystem (z. B.
len RNA-Polymerase und der Ge- det, da sie regelmäßig aktualisiert durch die Veränderung eines Anti-
Foto: freshidea/stock.adobe.com
samtgröße der viralen Population. wird und einfach in ihrer Anwen- körperepitops) auswirken können.
Ausgehend von einem monophyleti- dung ist (4, 5). Inwieweit bestimmte Mutationen
schen Ursprung wurde die geneti- Zu einem gewissen Teil ist die ge- die Fitness des Virus beeinflussen
sche Diversifizierung von SARS- netische Fluktuation von RNA-Vi- und ihm so eine erfolgreiche An-
CoV-2 begünstigt durch die explosi- ren immer zufallsbedingt. Im Rah- passung an den Selektionsdruck in
A 460 Deutsches Ärzteblatt | Jg. 118 | Heft 9 | 5. März 2021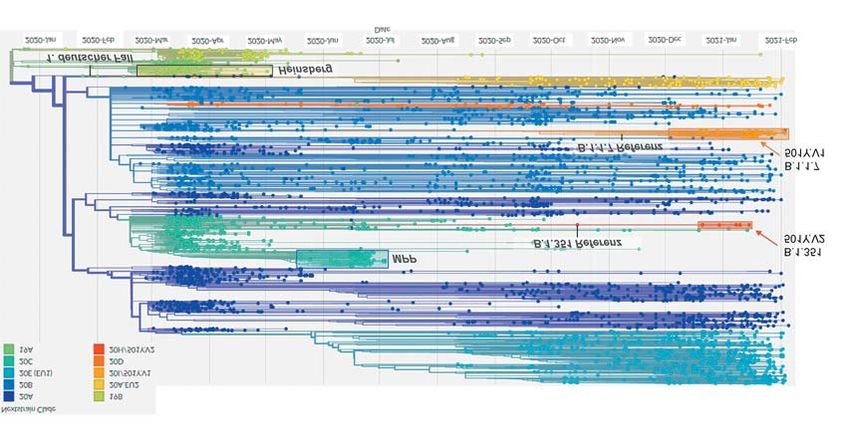
MEDIZINREPORT
der menschlichen Population er- Änderungen im Spike-Protein kön- barkeit des Erregers auswirken. Je
möglichen, wird seit Beginn der nen sich deshalb auf die Infektiosi- nachdem, wie deutlich die Hinwei-
Pandemie intensiv erforscht. Von tät des Virus sowie auf die Antikör- se auf derart veränderte Erreger-
besonderem Interesse sind in die- perneutralisation auswirken (3). eigenschaften sind, werden solche
sem Zusammenhang Veränderun- Ein erstes Beispiel einer im Sinne Varianten als Variante unter Beob-
gen des Spike-Rezeptorbindepro- der Virusverbreitung erfolgreichen achtung (variant under investiga-
teins. Hierbei handelt es sich um Mutation innerhalb des Spike-Pro- tion, VUI) oder besorgniserregende
ein 1 273 Aminosäuren umfassen- teins ist der Aminosäureaustausch Variante (variant of concern, VOC)
des Protein, das aus 2 Untereinhei- D614G, welcher während der ersten bezeichnet. Die WHO listet derzeit
ten besteht, die durch proteolyti- Pandemiewelle im Frühjahr 2020 3 VOCs: B.1.1.7, B.1.351 und P.1
sche Spaltung eines linearen Vor- erworben wurde. Aufgrund einer (Tabelle).
läufers entstehen (Grafik 1A). Konformationsänderung des Spike-
Das Spike-Protein ist für die Ver- Proteintrimers sind Viren des B.1.1.7 [501Y.V1; VOC 202012/01]
mehrungsfähigkeit des Virus essen- 614G-Genotyps infektiöser als der Im Dezember 2020 wurde aus
ziell, da es den Eintritt in die D614-Ursprungsgenotyp (9–11). Großbritannien über die zunehmen-
menschliche Wirtszelle vermittelt, Der 614G-Genotyp geht mit höhe- de Ausbreitung der neuartigen
wo die eigentliche Replikation des rer Viruslast und stärkerer Übertrag- SARS-CoV-2-Linie B.1.1.7 (auch:
Virus stattfindet. Die S1-Unterein- barkeit einher. Er herrscht mittler- 20I/501Y.V1) mit besorgniserre-
heit trägt die N-terminale Domäne weile weltweit vor, während Viren genden Eigenschaften hinsichtlich
(NTD) und die rezeptorbindende des ursprünglichen Genotyps D614 Infektiosität und Ausbreitung be-
Domäne (RBD), welche die Bin- fast gar nicht mehr zirkulieren (12). richtet (5, 13). Viren der Linie
dung an das zelluläre ACE2-Rezep- B.1.1.7 weisen 23 Polymorphismen
torprotein vermittelt (Grafik 1B); Besorgniserregende Varianten auf, von denen 17 in Aminosäure-
anschließend erfolgt über die Molekulare Surveillance ermög- änderungen der Virusproteine resul-
S2-Untereinheit die Fusion mit der licht die Beobachtung und Feindif- tieren, die vorwiegend das Spike-
Wirtszellmembran. ferenzierung zirkulierender SARS- Protein betreffen (Grafik 1). Ei-
Das Spike-Protein ist gleichzei- CoV-2-Varianten. Besondere Auf- ne charakteristische Deletion im
tig die Zielstruktur neutralisieren- merksamkeit gebührt hierbei neuen Spike-kodierenden S-Gen (S del
der (protektiver) Antikörper, die Varianten mit Mutationen, die sich 69/70) verursachte falsch-negative
insbesondere auf Regionen der auf die Übertragbarkeit, Immun- Nachweise des viralen S-Gens mit-
RBD und der NTD abzielen (7, 8). kontrolle, Virulenz oder Nachweis- tels bestimmter Multiplex-PCR-
Systeme. Dieser S-Gen-Ausfall war
es, der initial die Aufmerksamkeit
TABELLE
auf die Virusvariante lenkte und so
Variants of Concern von SARS-CoV-2 die frühe Erkennung dieser neuarti-
gen Viruslinie in Großbritannien
B.1.1.7 B.1.351 P.1
20I/501Y.V1 20H/501Y.V2 20J/501Y.V3 sowie zeitnahe Analysen zu ihrer
VOC 202012/01 VOC 202012/02 VOC 202101/03 Verbreitung anhand von klinisch-
Region des ersten Nachweises Großbritannien Südafrika Brasilien diagnostischen Labordaten ermög-
lichte.
Mutationen im Spike-Protein ΔH69/ΔV70, ΔY144, L18F, D80A, D215G, L18F, T20N, P26S,
(RBD) N501Y, A570D, P681H, R246I, K417N, E484K, D138Y, R190S, K417T, Die B.1.1.7-Variante hat sich
T716I, S982A, D1118H N501Y, A701V E484K, N501Y, H655Y, mittlerweile zur vorherrschenden
T1027I, V1176F Variante in Großbritannien entwi-
Transmissibilität ↑ ↑ ungeklärt ckelt und wurde bislang (Stand 17.
Rt (effektive Reproduktionszahl) ↑ (16,18) (↑) (36) ungeklärt Februar 2021) in 88 Ländern nach-
Secondary Attack Rate ↑ (17) ungeklärt ungeklärt
gewiesen, einschließlich der Bun-
desrepublik Deutschland. Auch in
In vitro Daten 501Y: 501Y & 484K: 501Y & 484K:
↑ ACE2 ↑↑↑ ACE2 ↑↑↑ ACE2 anderen Ländern wird eine rasche
Rezeptoraffinität (23, 24) Rezeptoraffinität (23) Rezeptoraffinität (22) Verbreitung mit Entwicklung hin zur
Immune escape Ausreichende Neutralisa- ↓ Suszeptibilität ↓ Suszeptibilität erwar- Prädominanz beobachtet (14, 15).
tion durch Plasma von gegen Geimpften-/ tet, da ähnliche Muta- Ursächlich für die erfolgreiche
Genesenen und Geimpf- Konvaleszentenplas- tionen vorliegen wie in Verbreitung von B.1.1.7 scheint ei-
ten (26, 37) ma (25, 38, 39) B.1.351
↓↓ Suszeptibilität ne im Vergleich zu anderen SARS-
(Resistenz) in vitro ge- CoV-2-Varianten leichtere Über-
gen bestimmte mono- tragbarkeit zu sein: Die Zahl von
klonale Antikörper (25)
Hinweise auf COVID-19-Fällen, bei denen im
↓ Impfstoffeffektivität Rahmen der PCR-Diagnostik ein
(28)
S-Gen-Ausfall berichtet wurde (als
Virulenz ↑ Fallsterblichkeit (20, 21) ungeklärt ungeklärt proxy für diese Variante), stieg ra-
scher an als die von Fällen ohne
Deutsches Ärzteblatt | Jg. 118 | Heft 9 | 5. März 2021 A 461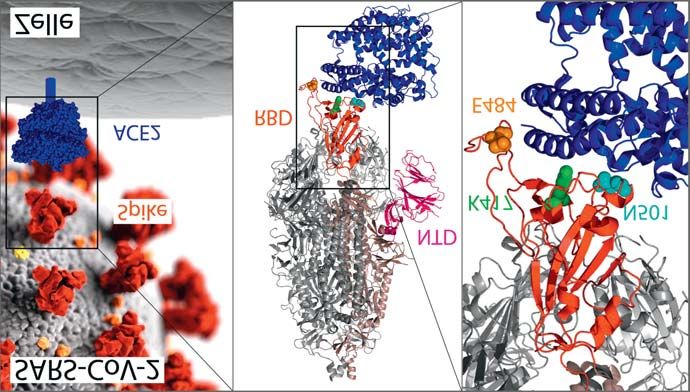
MEDIZINREPORT
GRAFIK 1A VOC in Südafrika berichtet, ausge-
hend von der Provinz Ostkap (22).
Spike-Protein-Gen mit variantenspezifischen Mutationen Auch diese Linie, B.1.351 (auch
(adaptiert von Wang et al. [25] mit Erlaubnis der Autoren)
20H/501Y.V2), entwickelte sich
rasch zur lokal vorherrschenden Va-
riante, weshalb eine erhöhte Trans-
missibilität vermutet wird.
Sie ist durch 9 Aminosäureände-
rungen gekennzeichnet, von denen
8 das Spike-Protein betreffen (Gra-
fik 1); hierzu gehört die von B.1.1.7
bekannte, jedoch unabhängig er-
Im für das Spike-Protein kodierenden Abschnitt des SARS-CoV-2-Genoms zeigen die Varianten B.1.1.7 (oben) und
worbene N501Y-Mutation, die für
B.1.351 (unten) charakteristische Mutationen.
NTD: N-terminale Domäne; RBD: rezeptorbindende Domäne; SD1/2: Subdomäne 1/2; FP: Fusionspeptid; HR1/2: Hep-
eine erleichterte Bindung an
tadenmuster 1/2; CH: Zentrale Helix; CD: Verbindungsdomäne; CT: zytoplasmatische Domäne. menschliche Wirtszellen verant-
wortlich gemacht wird (23, 24). Zu-
sätzlich zum N501Y-Polymorphis-
GRAFIK 1B
mus finden sich weitere Mutationen
in der RBD (K417N und E484K)
und zahlreiche Polymorphismen in
der NTD des Spike-Proteins. Beide
Domänen tragen Epitope für neu-
tralisierende Antikörper, weshalb
sie entscheidende Strukturen so-
wohl für die Immunantwort als
auch die Vakzinierung und Antikör-
Erstellt unter Verwendung der PyMOL 3D-Grafiksoftware (pymol.org).
pertherapie darstellen. Für den Ami-
nosäureaustausch E484K ist bekannt,
dass er Resistenz gegen bestimmte
monoklonale Antikörper vermittelt
(25–27).
Vorab veröffentlichte In-vitro-
Studien implizierten zudem eine ver-
ringerte Aktivität von Rekonvales-
zenten- beziehungsweise Geimpf-
tenplasma gegen Viren, die diese
Lokalisation der B.1.1.7- bzw. B.1.351-typischen RBD-Mutationen im SARS-CoV-2-Spike-Protein. 3 Mutationen tragen. Darüber hinaus
gibt es Meldungen, nach denen in
Impfstoffstudien der Phase III in
S-Gen-Ausfall (16). Kontaktnach- B.1.1.7-Variante zum Anstieg der Südafrika eine verringerte Effektivi-
verfolgungsdaten demonstrierten Fallzahl beigetragen hat, zeigen die tät der Vakzine gefunden wurde, was
unter den Kontaktpersonen von mit möglicherweise weitreichenden Fol- auf die dortige Zirkulation der
B.1.1.7 Infizierten eine höhere Se- gen der Ausbreitung einer hoch- B.1.351-Linie zurückgeführt wird
condary Attack Rate (1361 Infi- transmissiblen Variante von SARS- (28, 29). Es ist also möglich, dass
zierte pro 9228 Kontaktpersonen CoV-2 auf. Gerade weil von einer sich die B.1.351-Linie so rasch aus-
[15 %]) im Vergleich zu Kontakt- noch höheren Transmissibilität aus- gebreitet hat, weil sie über eine in-
personen, die mit anderen Viruslini- gegangen werden muss, sind die trinsisch höhere Transmissibilität als
en infizierten waren (1244 pro Maßnahmen zur Viruseindämmung der ursprüngliche Genotyp verfügt
11269 [11 %]) (17). Darüber hinaus (Containment) wichtig. Dies betont (23, 24) und/oder weil sie partiell
deuten unterschiedliche Modellie- die hohe Bedeutung einer engma- Mechanismen der menschlichen Im-
rungsrechnungen auf eine um das schigen Surveillance zur frühzeiti- munantwort entkommen kann. Sie
rund 1,5-fach erhöhte Reproduk- gen Detektion und molekularen könnte daher eine Grundlage für die
tionszahl der B.1.1.7-Variante hin Charakterisierung bekannter sowie Entstehung sogenannter Immune-
(15, 16, 18). Während für B.1.1.7- auch neuartiger Viruslinien. Escape-Varianten darstellen.
Infektionen initial eine unveränder-
te Klinik angenommen wurde, deu- B.1.351 [501Y.V2] P.1 [501Y.V3]
ten bei begrenzter Studienlage erste Nahezu zeitgleich mit Berichten Im brasilianischen Bundesstaat Ama-
Daten auf eine erhöhte Fallsterb- über die neue Variante aus Großbri- zonas nehmen derzeit Infektionen
lichkeit hin (19–21). Die Erfah- tannien wurde im Dezember 2020 mit einer weiteren SARS-CoV-2-
rungen aus Großbritannien, wo die über die Ausbreitung einer weiteren Variante rasch zu, die als P.1 bzw.
A 462 Deutsches Ärzteblatt | Jg. 118 | Heft 9 | 5. März 2021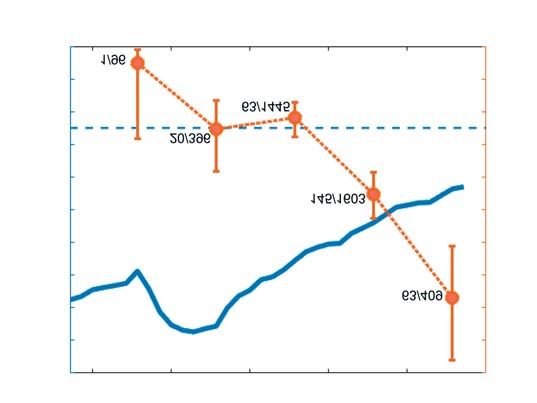
MEDIZINREPORT 501Y.V.3 bezeichnet wird. Sie weist Die zellulären und virologischen das Genom der jeweiligen SARS- ebenfalls eine Reihe von Spike-Pro- Determinanten, die einen VOC- CoV-2-Variante zusammen mit den tein-Polymorphismen auf, darunter Phänotyp bestimmen, herauszuar- klinisch-epidemiologischen Infor- auch 3 RBD-Mutationen an Positio- beiten, ist Gegenstand intensiver mationen der jeweiligen Fälle aus- nen, die auch in der B.1.351-Linie Forschungsanstrengungen. Neben gewertet werden. Somit können in verändert sind (K417, E484, N501). vergleichenden Analysen in Zell- kurzer Zeit Zusammenhänge von Aufgrund des Genotyps wird auch kultur-, Organoid- und Tiermodel- Viruslinien mit Krankheitsverläufen für diese Variante eine erhöhte len stehen hierfür klinische Beob- erkannt werden. Transmissibilität beziehungsweise achtungen (z. B. Manifestationsfor- Basierend auf den erhobenen Ge- verringerte Effektivität neutralisie- men, Dauer, Menge der Virusaus- nomdaten und Daten, die in Reposi- render Antikörper diskutiert (30). scheidung), syndrombasierte und torien wie GISAID eingestellt wer- Die Mechanismen, die VOCs ei- integrierte Molekulare Surveillance den, liegen derzeit (Stand: 11. Febru- ne überdurchschnittliche Übertrag- zur Erfassung der Prävalenz und ar 2021) Informationen zu insgesamt barkeit verleihen, sind derzeit nur Verbreitung von Varianten sowie 5 875 deutschen (2020: 4 552; 2021: im Ansatz verstanden. Sie scheinen der Schwere der Infektion und pa- 1 323) SARS-CoV-2-Gesamtgenom- jedoch durch gemeinsame Poly- thologische Untersuchungen zur sequenzen vor. Grafik 2A gibt einen morphismen im viralen S-Protein Erkennung von Unterschieden in Gesamtüberblick über die Verwandt- wie N501Y getrieben zu sein. Die- der klinisch-pathologischen Mani- schaft der in Deutschland seit Beginn se verstärken die Bindung an das festation zur Verfügung. der Pandemie sequenzierten SARS- ACE2-Rezeptorprotein und geben CoV-2-Genome. Es wird ersichtlich, einen Hinweis auf konvergente Aktuelle Situation in Deutschland dass eine Diversifikation erfolgt ist Anpassungsmechanismen des ur- Das Robert Koch-Institut (RKI) und und im Zeitverlauf die Häufigkeits- sprünglich zoonotischen Virus (23, das Konsiliarlabor für Coronaviren verteilung der unterschiedlichen Kla- 24). Denkbar ist, dass die spezi- der Charité – Universitätsmedizin den variiert. fisch in VOCs gefundenen Amino- Berlin führen seit Beginn der Pande- Es ist wichtig zu erwähnen, dass säureaustausche zu einer Verringe- mie Genomsequenzierungen von die sequenzierten Viren keine reprä- rung der minimalen Infektionsdo- SARS-CoV-2- durch, unter anderem sentative Auswahl aller in Deutsch- sis führen und die infektiöse Form im Rahmen der Integrierten Mo- land zirkulierenden Viren darstellen: des Virus stabilisieren, den zellulä- lekularen Surveillance (IMS) für Aufwendige Laboruntersuchungen ren Tropismus beeinflussen oder SARS-CoV-2. Die besondere Be- wie die Gesamtgenomsequenzierung die virale Fitness in anderer Form deutung der IMS liegt in der Tatsa- kommen vor allem in besonderen Si- erhöhen. che, dass die Informationen über tuationen zum Einsatz, etwa bei Aus- GRAFIK 2A Deutsche SARS-CoV-2-Gesamtgenomsequenzen seit Beginn der Pandemie Die Analyse erfolgte unter Verwendung der Augur- und Auspice-Pipelines am RKI (40, 41). Die berücksichtigten Genomsequenzen sind als Punkte in Relation zum Entnahmezeitpunkt der entsprechenden Probe (x-Achse) hervorgeho- ben, während die entsprechende Kladenzuordnung gemäß Nextstrain-Nomenklatur farbkodiert ist. Die Position der SARS-CoV-2-Sequenz des ersten deutschen COVID-19-Falls und Sequenzen aus großen Ausbrüchen sind durch Pfeile gekennzeichnet, ebenso wie die Cluster der VOC- Linien B.1.1.7 und B.1.351 (20I/501Y.V1 und 20H/501Y.V2 gemäß Nextstrain-Nomenklatur). Aus Datenschutzgründen sind nicht alle verfügba- ren Sequenzen dargestellt (MPP: fleischverarbeitender Betrieb). Deutsches Ärzteblatt | Jg. 118 | Heft 9 | 5. März 2021 A 463
MEDIZINREPORT
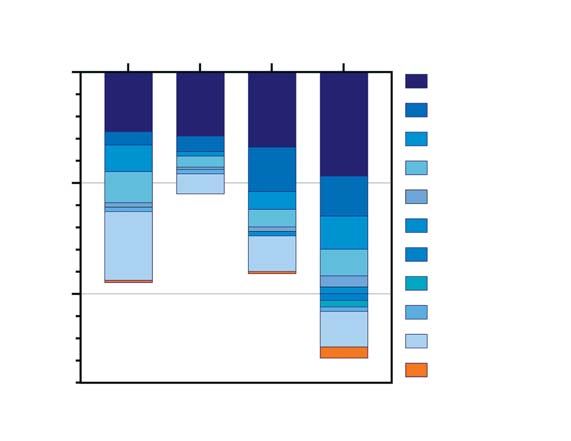
GRAFIK 2B 2020 ausgehend von Urlaubsrück-
kehrern aus Spanien ausbreitete
Linienverteilung der in IMS sequenzierten SARS-CoV-2, KW 50/2020–KW4/2021 und seitdem in ganz Europa vor-
herrscht. Es gibt bislang keine pu-
B.1.1.7
blizierten Hinweise darauf, dass
andere Linien sich B.1.177-Viren in ihren biologi-
B.1.1.74 schen Eigenschaften beziehungs-
100
B.1.9.4
weise ihrer Transmissibilität von
anderen SARS-CoV-2-Linien unter-
Sequenzzahl
B.1.88
scheiden (31).
B.1.1.238 Eine ebenfalls hohe Prävalenz
B.1 zeigen Viren der in ganz Europa
50 weitverbreiteten Linien B.1.221 und
B.1.258
B.1.160. Der Anteil der besorgniser-
B.1.160
regenden Variante B.1.1.7 an der
B.1.221 Gesamtzahl der Sequenzen der zu-
0 B.1.177 fällig ausgewählten IMS-SC2-Pro-
ben lag im Erhebungszeitraum noch
50–51 52–53 1–2 3–4
Kalenderwoche (Entnahme)
auf niedrigem Niveau, zeigte jedoch
einen deutlichen Aufwärtstrend mit
einem relativen Anteil von 3,9 %
Im Gegensatz zum in 2A dargestellten Datensatz basieren die hier erhobenen und ausgewerte- in KW 3–4.
ten Sequenzdaten auf einer Zufallsstichprobe, für die eine geringere Verzerrung (z. B. im Sinne Damit stehen die IMS-SC2-Sur-
der Überrepräsentation von VOCs) anzunehmen ist. Dargestellt ist die Anzahl der 10 am häu-
veillance-Ergebnisse im Einklang
figsten nachgewiesenen SARS-CoV-2-Linien an allen sequenzierten SARS-CoV-2-positiven
Proben aus dem IMS-SC2-Labornetzwerk. mit anderen Erhebungen des RKI:
Eine Ad-hoc-Analyse fand in KW 4
einen Anteil B.1.1.7-positiver PCR-
bruchsgeschehen, bei schweren klini- schen Breite Deutschlands erhoben. Ergebnisse von 5,6 %, der an-
schen Verläufen oder dem Verdacht Daraus resultiert ein Datensatz, der schließend rapide auf 22 % anstieg
auf das Vorliegen einer VOC. Daher das Infektionsgeschehen mit gerin- (32, 33). Die Testzahlerfassung des
unterliegen viele der in Sequenzda- geren Verzerrungen hinsichtlich der RKI demonstrierte ebenfalls einen
tenbanken wie GISAID hinterlegten Probenauswahl repräsentiert, als es VOC-Anteil von 5 % in KW 4, der
Datensätze einer Verzerrung, in der bei Proben der Fall ist, die aus diag- in KW 5 auf 12 % und in KW 6 auf
sehr homogene Datensätze aus ein- nostischen Zwecken zur Sequenzie- 22,8 % anstieg. Es handelt sich aber
zelnen Untersuchungen beziehungs- rung ausgewählt wurden. nicht um eine zufällige Stichprobe
weise mittlerweile auch VOC-Se- Zum Zweck der Surveillance (32, 33).
quenzen überrepräsentiert sind. senden derzeit (Stand 17. Februar Seit Januar 2021 ist der Deutsche
2021) 17 über Deutschland verteilte Elektronische Sequenzdaten-Hub
Molekulare Surveillance Diagnostiklabore wöchentlich zu- (DESH) aktiv, über den sequenzie-
Das Projekt „Integrierte Molekula- fällig ausgewählte Proben zur Ge- rende Labore SARS-CoV-2-Se-
re Surveillance für SARS-CoV-2 samtgenomsequenzierung an das quenzdaten an das RKI übermitteln
(IMS-SC2)“ an RKI und Charité RKI. Darüber hinaus erfolgen fo- (34). Diese technische Plattform,
wurde etabliert, um (i) die geneti- kussierte Untersuchungen auf Ver- welche im Rahmen der Corona-
sche Variabilität und Evolution von anlassung des Öffentlichen Ge- Surveillanceverordnung realisiert
SARS-CoV-2 in Deutschland konti- sundheitsdienstes, ebenfalls breit wurde und die Grundlage und Mo-
nuierlich zu überwachen, (ii) einen über Deutschland verteilt. Seit De- dell auch für die IMS anderer Er-
differenzierten, durch molekularge- zember 2020 sind so im Rahmen reger sein kann, ermöglicht die
netische Analysen unterstützten des IMS-SC2-Projektes am RKI deutschlandweite Zusammenfüh-
Einblick in das Auftreten sowie die 658 SARS-CoV-2-Genome sequen- rung von Sequenzdaten und wird
Ausbreitungs- und die Transmissi- ziert worden, darunter 440 im Rah- kontinuierlich weiterentwickelt. Ei-
onsdynamiken des Virus zu gewin- men der Surveillance von zufällig ne Analyse der an DESH übermit-
nen,und (iii) einen Zusammenhang ausgewählten Proben. telten Daten von Zufallsstichpro-
zwischen Infektionen mit bestimm- Grafik 2A zeigt für die Kalender- ben aus sequenzierenden Laboren
ten Virusvarianten und klinisch-epi- wochen (KW) 50/2020 bis 4/2021 in ganz Deutschland sowie der
demiologischen Erscheinungsbil- die Verteilung dieser Zufallsstich- IMS-SC2-Sequenzdaten zufällig se-
dern zu erkennen. probe auf die unterschiedlichen lektierter SARS-CoV-2-Proben de-
Dementsprechend wird ein großer SARS-CoV-2-Linien im Zeitver- monstrierte in KW 3 einen Anteil
Anteil der Sequenzen anhand von zu- lauf. Derzeit entfällt der größte Teil von 4,4 % für B.1.1.7-Sequenzen
fällig ausgewählten SARS-CoV-2- der zirkulierenden Viren auf die Li- an den in Deutschland sequenzier-
positiven Proben aus der geografi- nie B.1.177, die sich im Sommer ten SARS-CoV-2-Genomen, der in
A 464 Deutsches Ärzteblatt | Jg. 118 | Heft 9 | 5. März 2021MEDIZINREPORT
GRAFIK 2C treten von Virusvarianten, die par-
tiell oder komplett resistent gegen
Anteil der VOC B.1.1.7 an sequenzierten SARS-CoV-2-Genomen neutralisierende Antikörper oder
Linke Y-Achse:
deutschlandweite Rekonvaleszentenseren sind, deutet
200 20
7-Tage-Inzidenz darauf hin, dass sich SARS-CoV-2
(gemeldete SARS- 180 18 an den herrschenden Immunselek-
CoV-2-Neuinfektio- tionsdruck anpassen kann. Eine sol-
nen/100 000 Ein- 160 16
7-Tage-Inzidenz von SARS-CoV-2
che Antigendrift könnte die Wirk-
wohner/7 Tage);
(Fälle/100 000 Einwohner)
140 14 samkeit von Impfstoffen negativ be-
rechte Y-Achse:
Anteil B.1.117 (%)
prozentualer 120 12 einflussen und würde unter Umstän-
B.1.1.7-Anteil unter den eine Anpassung der Impfstoff-
100 10
3 949 zufällig aus- komposition erfordern, ähnlich wie
gewählten SARS- 80 8 dies für Influenza schon der Fall
CoV-2-Proben ist (35). Hieraus ergibt sich die
60 6
(DESH oder IMS- Notwendigkeit einer engmaschigen
SC2); für diese sind 40 4 Überwachung der Evolution von
auch absolute Zah-
len (Anzahl B.1.1.7 20 2 SARS-CoV-2. Die Coronavirus-
Sequenzen/Ge- Surveillanceverordnung (CorSurV)
0 0
samtzahl SARS- 3.1.21 10.1.21 17.1.21 24.1.21 31.1.21 7.2.21 vom 19. Januar 2021 unterstützt zu
CoV-2-Sequenzen) Datum diesem Zweck eine erhebliche Stei-
gezeigt (33, 34). gerung der SARS-CoV-2-Gesamt-
genomsequenzierungen mit dem
Ziel der Sequenzierung von 5–10 %
KW 4 auf 9,0 % und in KW 5 auf 3 unterschiedlichen Weltregionen aller SARS-CoV-2-positiven Proben
15,4 % angestiegen ist (33). ist ein Indiz für konvergente Evolu- in Deutschland (je nach Inzidenz).
Grafik 2C zeigt für die Kalender- tion. Quantitative und longitudinale
wochen 1–5/2021 den steigenden Anlass zur Besorgnis stellt derzeit Analysen auf Basis der in DESH er-
B.1.1.7-Anteil an den Gesamtge- die um rund 50 % leichter übertrag- fassten Genomsequenzen erlauben
nomsequenzen in DESH bei insge- bare Linie B.1.1.7 dar (16,18). Daten die Etablierung eines Frühwarnsys-
samt noch sinkender 7-Tage-Neuin- aus der Integrierten Molekularen tems zur Erkennung von sich über-
zidenz (Summe aller Varianten). Surveillance von RKI und Konsiliar- proportional ausbreitenden bekann-
Viren der Linie B.1.351 werden labor für Coronaviren an der Charité ten, aber auch neuen Varianten, die
mit einem Anteil von 1,23 % (Stand sowie aus groß angelegten multizen- dann gezielt beobachtet und analy-
17. Februar 2021) in Sequenzdaten trischen Ad-hoc-Analysen demons- siert werden können. Diese Surveil-
gefunden, die via DESH an das trieren, dass der Anteil der durch lance leistet somit einen wertvollen
RKI übermittelt werden. Viren der B.1.1.7 verursachten Neuinfektionen Informationszugewinn für die Pan-
Linie P.1 wurden bislang weder in Ende Januar 2021 bei knapp 6 % und demiekontrolle. Unabhängig von
der IMS-SC2 noch in den via 2 Wochen später bei geografischer SARS-CoV-2 ist die jetzt etablierte
DESH übermittelten Daten detek- Diversität durchschnittlich bereits IMS ein Modell, das künftig auch
tiert, sind in Deutschland aber be- bei 22 % lag (32, 33). für weitere Infektionserreger etab-
reits zumindest einmal nachgewie- Somit dominiert diese Variante liert und genutzt werden sollte, um
sen worden (33). das Infektionsgeschehen in Deutsch- den Gesundheitsschutz in Deutsch-
land noch nicht komplett, aber eine land weiter zu verbessern.
Zusammenfassende Bewertung weitere Ausbreitung und ein Einfluss RKI-Autorengruppe „SARS-CoV-2
Mit zunehmender Verbreitung und auf die Transmission muss erwartet Integrierte Molekulare Surveillance“
Populationsgröße von SARS-CoV-2 werden. Die Ad-hoc-Analysen wer-
steigt die Wahrscheinlichkeit des den in den kommenden Wochen wie- Korrespondenz:
PD Dr. rer. nat. Thorsten Wolff
Auftretens phänotypisch-relevanter derholt werden, um den zeitlichen Robert Koch-Institut, Fachgebiet 17: Influenza-
Mutationen im Virusgenom. Die Verlauf des VOC-Anteils in Deutsch- viren und weitere Viren des Respirationstraktes,
daraus resultierende hohe geneti- land zu verfolgen. Wie effektiv die E-Mail: wolfft@rki.de
sche Diversität ermöglicht eine derzeitigen Maßnahmen die Aus- Interessenkonflikte: Die Autoren geben an, dass
Selektion angepasster Virusvarian- breitung der VOCs begrenzen, wird keine Interessenkonflikte bestehen.
ten, deren Fitness zum Beispiel sich aus den Surveillancedaten-Er-
aufgrund leichterer Übertragbarkeit gebnissen der nächsten Tage und Der Beitrag unterlag keinem Peer-Review-Verfahren.
oder geringerer Suszeptibilität ge- Wochen ablesen lassen.
gen die menschliche Immunant- Sicher ist aber, dass aufgrund der Eine vollständige Liste der Autoren:
http://daebl.de/PC74
wort erhöht ist. Das unabhängige höheren Transmission die Eindäm-
Auftreten von VOCs mit ähnlichen mung der VOC B.1.1.7 schwieriger
Literatur im Internet:
oder gar denselben Mutationen ist als die der bisher dominierenden www.aerzteblatt.de/lit0921
(S-N501Y; K417N/T; E484K) in SARS-CoV-2-Varianten. Das Auf- oder über QR-Code.
A 466 Deutsches Ärzteblatt | Jg. 118 | Heft 9 | 5. März 2021MEDIZINREPORT
Zusatzmaterial Heft 9/2021, zu:
SARS-CoV-2-Varianten
Evolution im Zeitraffer
Für den Fortgang des pandemischen Geschehens spielen Fragen zu Eigenschaften, Verbreitung und
Bedeutung mutierter Varianten von SARS-CoV-2 eine große Rolle. Die molekulare Surveillance erfolgt
am Robert Koch-Institut unter anderem mittels Gesamtgenomsequenzierungen.
Literatur 2-variant-variant-of-concern-20201201 (last 10.1101/2020.12.21.20248640.
1. Cui J, Li F, Shi ZL: Origin and evolution of accessed on 26 January 2021). 23. Zahradník J, Marciano S, Shemesh M, et
pathogenic coronaviruses. Nat Rev Micro- 14. Borges, V, Sousa C, Menezes L, et al.: Tra- al.: SARS-CoV-2 RBD in vitro evolution fol-
biol 2019; 17: 181–92. cking SARS-CoV-2 VOC 202012/01 (linea- lows contagious mutation spread, yet gene-
2. Mallapaty S, Maxmen A, Callaway E: „Major ge B.1.1.7) dissemination in Portugal: in- rates an able infection inhibitor. bioRxiv 8.
stones unturned“: COVID origin search sights from nationwide RT-PCR Spike gene Januar 2021; doi:
must continue after WHO report, say scien- drop out data. Virological.org 2021; 10.1101/2021.01.06.425392.
tists. Nature 10. Februar 2021. https://virological.org/t/tracking-sars-cov- 24. Starr TN, Greaney AJ, Hilton SK, et al.:
2-voc-202012–01-lineage-b-1–1–7-dissemi- Deep Mutational Scanning of SARS-CoV-2
3. Coronaviridae Study Group of the Interna- nation-in-portugal-insights-from-nationwide-
tional Committee on Taxonomy of Viruses: Receptor Binding Domain Reveals Con-
rt-pcr-spike-gene-drop-out-data/600 (last straints on Folding and ACE2 Binding. Cell
The species Severe acute respiratory syn- accessed on 26 January 2021).
drome-related coronavirus: classifying 2020; 182: 1295–310.
2019-nCoV and naming it SARS-CoV-2. 15. Statens Serum Institut: Nyt kontakttal for vi- 25. Wang P, LiuL, Iketani S, et al.: Antibody Re-
Nat Microbiol 2020; 5: 536–44. rusvariant B.1.1.7. 2021; https://www.ssi.dk/ sistance of SARS-CoV-2 Variants B.1.351
aktuelt/nyheder/2021/nyt-kontakttal-for- and B.1.1.7. bioRxiv 26. Januar 2021; doi:
4. Alm, E, Broberg EK, Connor T, et al.: Geo- virusvariant-b117 (last accessed on 26 Ja-
graphical and temporal distribution of 10.1101/2021.01.25.428137.
nuary 2021).
SARS-CoV-2 clades in the WHO European 26. Wu K, Werner AP, Moliva JI, et al.:
Region, January to June 2020. Euro Sur- 16. Volz E, Mishra S, Chand M, et al.: Trans- mRNA-1273 vaccine induces neutralizing
veill 2020; 25 (32): 2001410. mission of SARS-CoV-2 Lineage B.1.1.7 in antibodies against spike mutants from glo-
England: Insights from linking epidemiologi- bal SARS-CoV-2 variants. bioRxiv 25. Janu-
5. Rambaut A, Loman N, Pybus O, et al.: Pre- cal and genetic data. medRxiv, 4. Januar
liminary genomic characterisation of an ar 2021; doi: 10.1101/2021.01.25.427948.
2020; doi: 10.1101/2020.12.30.20249034.
emergent SARS-CoV-2 lineage in the UK 27. Greaney AJ, Loes AN, Crawford KHD, et
defined by a novel set of spike mutations. 17. Public Health England: Investigation of no- al.: Comprehensive mapping of mutations
Virological 2020; https://virological.org/t/pre vel SARS-CoV-2 variant. Variant of Concern to the SARS-CoV-2 receptor-binding do-
liminary-genomic-characterisation-of-an- 202012/01. Technical briefing 3 2021; main that affect recognition by polyclonal
emergent-sars-cov-2-lineage-in-the-uk-defi https://assets.publishing.service.gov.uk/go human serum antibodies. bioRxiv 4. Januar
ned-by-a-novel-set-of-spike-mutations/563 vernment/uploads/system/uploads/attach 2020; 10.1101/2020.12.31.425021.
(last accessed on 26 January 2021). ment_data/file/959360/Variant_of_Con
cern_VOC_202012_01_Technical_Brie 28. Madhi SA, Baillie V, Cutland CL, et al.:
6. Rambaut A, Posada D, Crandall KA, fing_3.pdf. Safety and efficacy of the ChAdOx1
Holmes EC: The causes and consequences nCoV-19 (AZD1222) Covid-19 vaccine
of HIV evolution. Nat Rev Genet 2004; 5: 18. Vöhringer H, Sinnott M, Amato R, et al.: against the B.1.351 variant in South Africa.
52–61. Lineage-specific growth of SARS-CoV-2 medRxiv 12. Februar 2021; doi:
B.1.1.7 during the English national lock- 10.1101/2021.02.10.21251247.
7. Enjuanes L, Smerdou C, Castilla J, et al.: down. Virological.org 3. Dezember 2020;
Development of protection against corona- https://virological.org/t/lineage-specific- 29. Novavax-Impfstoff: Vor allem die südafrika-
virus induced diseases. A review. Adv Exp growth-of-sars-cov-2-b-1–1–7-during-the- nische Variante mindert die Schutzwirkung
Med 1995; Biol 380: 197–211. english-national-lockdown/575. gegen COVID-19. aerzteblatt.de 29. Januar
8. Liu L, Wang P, Nair MS, et al.: Potent neu- 2021; http://daebl.de/PE17.
19. European Centre for Disease Prevention
tralizing antibodies against multiple epito- and Control: Rapid increase of a SARS- 30. Faria NR, Claro IM, Candido D, et al.: Ge-
pes on SARS-CoV-2 spike. Nature 2020; CoV-2 variant with multiple spike protein nomic characterisation of an emergent
584: 450–6. mutations observed in the United Kingdom SARS-CoV-2 lineage in Manaus: prelimina-
9. Yurkovetskiy L, Wang X, Pascal KE, et al.: 20. Dezember 2020; https://www.ecdc. ry findings. virological.org 13. Januar 2021;
Structural and Functional Analysis of the europa.eu/sites/default/files/documents/ https://virological.org/t/genomic-characteri
D614G SARS-CoV-2 Spike Protein Variant. SARS-CoV-2-variant-multiple-spike-protein- sation-of-an-emergent-sars-cov-2-lineage-
Cell 2020; 183: 739–51. mutations-United-Kingdom.pdf. in-manaus-preliminary-findings/586 (last ac-
cessed on 26 January 2021).
10. Hou YJ, Chiba S, Halfmann P, et al.: SARS- 20. Davies NG, Jarvis CI, CMMID COVID-19
CoV-2 D614G variant exhibits efficient repli- Working Group, et al.: Increased hazard of 31. Hodcroft EB, Zuber M, Nadeau S, et al.:
cation ex vivo and transmission in vivo. death in community-tested cases of SARS- Emergence and spread of a SARS-CoV-2
Science 2020; 370: 1464–8. CoV-2 Variant of Concern 202012/01. variant through Europe in the summer of
medRxiv 3. Februar 2021; doi: 2020. medRxiv 27. November 2020; doi:
11. Plante, JA, et al.: Spike mutation D614G al- 10.1101/2020.10.25.20219063.
ters SARS-CoV-2 fitness. Nature 26 Okto- 10.1101/2021.02.01.21250959.
ber 2020; doi: 10.1038/s41586-020-2895-3. 21. New and Emerging Respiratory Virus 32. Robert Koch-Institut: Bericht zu Virusvarian-
Threats Advisory Group: Update note on ten von SARS-CoV-2 in Deutschland, ins-
12. Korber, B, Fischer WM, Gnanakaran S, et besondere zur Variant of Concern (VOC)
al.: Tracking Changes in SARS-CoV-2 B.1.1.7 severity 11. Februar 2021;
https://www.gov.uk/government/publicati B.1.1.7. 2021; https://www.rki.de/DE/Con
Spike: Evidence that D614G Increases In- tent/InfAZ/N/Neuartiges_Coronavirus/
fectivity of the COVID-19 Virus. Cell 2020; ons/nervtag-update-note-on-b117-severity-
11-february-2021. DESH/Berichte-VOC-tab.html (last acces-
182: 812–27. sed on 26 January 2021).
13. Public Health England: Investigation of no- 22. Tegally H, Wilkinson E, Giovanetti M, et al.:
Emergence and rapid spread of a new se- 33. Robert Koch-Institut: 2. Bericht zu Virusvari-
vel SARS-COV-2 variant: Variant of Con- anten von SARS-CoV-2 in Deutschland,
cern 202012/01. PHE Technical Briefing vere acute respiratory syndrome-related co-
ronavirus 2 (SARS-CoV-2) lineage with mul- insbesondere zur Variant of Concern (VOC)
2020; https://www.gov.uk/government/publi B.1.1.7. 2021; https://www.rki.de/DE/Con
cations/investigation-of-novel-sars-cov- tiple spike mutations in South Africa.
medRxiv 22. Dezember 2020; doi: tent/InfAZ/N/Neuartiges_Coronavirus/
A7 Deutsches Ärzteblatt | Jg. 118 | Heft 9 | 5. März 2021MEDIZINREPORT
DESH/DESH.html (last accessed on 26 Ja-
nuary 2021).
34. Robert Koch-Institut: Deutscher Elektroni-
scher Sequenzdaten-Hub (DESH) 2020.
35. Bieniasz, P: The case against delaying
SARS-CoV-2 mRNA vaccine boosting do-
ses. Clin Infect Dis 27. Januar 2021; doi:
10.1093/cid/ciab070.
36. Pearson CA, Russell TW, Davies NG, et al.:
Estimates of severity and transmissibility of
novel South Africa SARS-CoV-2 variant
501Y.V2. 2021; https://cmmid.github.io/to
pics/covid19/reports/sa-novel-variant/
2021_01_11_Transmissibility_and_
severity_of_501Y_V2_in_SA.pdf (last ac-
cessed on 26 January 2021).
37. Muik A, Wallisch AK, Sänger B, et al.: Neu-
tralization of SARS-CoV-2 lineage B.1.1.7
pseudovirus by BNT162b2 vaccine-elicited
human sera. bioRxiv 19. Januar 2021; doi:
10.1101/2021.01.18.426984.
38. Wibmer CK, Ayres F, Hermanus T, et al.:
SARS-CoV-2 501Y.V2 escapes neutraliza-
tion by South African COVID-19 donor plas-
ma. bioRxiv 19. Januar 2021; doi:
10.1101/2021.01.18.427166.
39. Cele S, Gazy I, Jackson L, et al.: Escape of
SARS-CoV-2 501Y.V2 variants from neutra-
lization by convalescent plasma. medRxiv
26. Januar 2021; doi:
10.1101/2021.01.26.21250224.
40. Hadfield J, Megill N, Bell SM, et al.: Next-
strain: real-time tracking of pathogen evolu-
tion. Bioinformatics 2018; 34: 4121–3.
41. Huddleston J, Hadfield J, Sibley TR, et al.:
Augur: a bioinformatics toolkit for phyloge-
netic analyses of human pathogens. Jour-
nal of Open Source Software 2021; 6 (57):
2906.
Deutsches Ärzteblatt | Jg. 118 | Heft 9 | 5. März 2021 A8MEDIZINREPORT
Zusatzmaterial Heft 9/2021, zu:
SARS-CoV-2-Varianten
Evolution im Zeitraffer
Für den Fortgang des pandemischen Geschehens spielen Fragen zu Eigenschaften, Verbreitung und
Bedeutung mutierter Varianten von SARS-CoV-2 eine große Rolle. Die molekulare Surveillance erfolgt
am Robert-Koch-Institut unter anderem mittels Gesamtgenomsequenzierungen.
Autoren Danksagung
Dr. med. Djin-Ye Oh M.Sc 1*, Die Autoren danken Prof. Dr. Christian Drosten und
2
Dr. rer. nat. Stefan Kröger *, Dr. Victor Corman für die fortwährende Unterstüt-
Dr. rer. nat. Marianne Wedde ,
1 zung des Labornetzwerkes sowie die enge und ver-
3 trauensvolle Zusammenarbeit im Bereich der virolo-
Felix Hartkopf ,
gischen Surveillance von SARS-CoV-2. Dank gilt
Dr. rer. nat. Matthias Budt1, den Autoren zufolge auch den derzeit 17 Laboren
2
Dr. rer. nat. Janna Seifried , des IMS-SC2-Labornetzwerkes, die durch die konti-
3
Dr. rer. nat. Aleksandar Radonic , nuierliche Bereitstellung von Probenmaterial die re-
4
Essia Belarbi PhD , gelmäßige Analyse von SARS-CoV-2-Genomen
3
Dr. rer. nat. Martin Hölzer , aus verschiedenen Regionen Deutschlands erst er-
Dr. rer. nat. Sindy Böttcher ,
1 möglichen. Darüber hinaus danken sie allen se-
4
Dr. rer. nat. Grit Schubert , quenzierenden Laboren für die Bereitstellung von
3 Genomsequenzdaten über die DESH-Plattform des
Sandra Kaiser M.Sc ,
2 RKI oder öffentliche Repositorien wie GISAID. Des
Teresa Domaszewska PhD , Weiteren danken die Autoren Dr. Ulrike Elgeti, Dr.
4
Andreas Sachse , Jana Eckert und Dr. Astrid Broschkowski für die kri-
3
Dr. rer. nat. Oliver Drechsel , tische Durchsicht des Manuskripts.P5 Systemmedi-
2
Romina Grajcar , zin von Infektionskrankheiten.
3
Dr. rer. nat. Matthew Huska ,
5
Lanxin Zhang M.Sc ,
6
Dr. rer. nat. Annika Brinkmann ,
3
Kyanoush Yahosseini PhD ,
3
PD Dr. med. Linus Grabenhenrich ,
2
Dr. med. Osamah Hamouda MPH ,
1
Dr. med. vet. Ralf Dürrwald ,
Prof. Dr. med. Walter Haas²,
4
Sébastien Calvignac-Spencer PhD ,
3
Torsten Semmler Dr. rer. nat. ,
6
Prof. Dr. med. Lars Schaade ,
1
Prof. Dr. med. Martin Mielke ,
5
Max von Kleist PhD, M.Sc ,
3
Dr. rer. nat. Stephan Fuchs ,
3
Prof. Dr. med. vet. Lothar H. Wieler ,
1
PD Dr. rer. nat. Thorsten Wolff
1
RKI, Abteilung 1, Infektionskrankheiten
2
RKI, Abteilung 3, Infektionsepidemiologie
3
RKI, MF, Methodenentwicklung und
Forschungsinfrastruktur
4
RKI, Projektgruppe 3, Epidemiologie
hochpathogener Erreger
5
RKI, Projektgruppe 5, Systemmedizin von
Infektionskrankheiten
6
RKI, ZBS, Zentrum für Biologische Gefahren und
Spezielle Pathogene
* Diese Autoren trugen gleichermaßen bei.
A9 Deutsches Ärzteblatt | Jg. 118 | Heft 9 | 5. März 2021Sie können auch lesen

















































