Homepage: www.kup.at/klinendokrinologie Online-Datenbank mit Autoren- und Stichwortsuche
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

Kleinwuchs – Differenzialdiagnose und therapeutische Optionen
Kapelari K
Journal für Klinische Endokrinologie und Stoffwechsel - Austrian
Journal of Clinical Endocrinology and Metabolism 2015; 8 (1), 5-14
Homepage:
www.kup.at/klinendokrinologie
Online-Datenbank mit Autoren- und Stichwortsuche
Austrian Journal of Clinical Endocrinology and Metabolism
Metabolism
Kleinwuchs – Differenzialdiagnose
und therapeutische Optionen
Kleinwuchs – Differenzialdiagnose
und therapeutische Optionen
K. Kapelari
Kurzfassung: Auffälligkeiten des Wachstums ei- zentilenkurve, dar. Für einige definierte Wachs- of auxological parameters including skeletal pro-
nes Kindes gehören zu den häufigsten Fragestel- tumsstörungen stehen Therapien zur Verfügung. portions, growth velocity, and calculation of fa-
lungen in der Pädiatrie. Die fachlich korrekte Beur- Über 40 Jahre lang stand zur Behandlung ledig- milial target height is requested to discriminate
teilung im individuellen Fall setzt eine gewissen- lich Wachstumshormon zur Verfügung, seit 1985 treatable from non-treatable growth disorders.
hafte Erfassung der auxologischen Verlaufspara- in rekombinanter Form und somit praktisch unbe- A carefully drawn growth chart is indispens-
meter, die profunde Kenntnis des physiologischen schränkt verfügbar. Seit der Markteinführung von able. Estimation of bone age based on an x-ray
Wachstumsverlaufes von Kindern und das Wissen rekombinantem IGF-1 ergeben sich neue thera- of the left hand and calculation of prospected fi-
über die Vielzahl von möglichen Wachstumsstö- peutische Optionen. nal height provide further issues to differenti-
rungen voraus. Das Wachstum von Kindern unter- ate between inborn and secondary growth fail-
liegt einer großen natürlichen Variationsbreite – Schlüsselwörter: Kleinwuchs, Wachstumsstö- ure. For more than forty years, classification of
ein Umstand, der die Abgrenzung von sog. Norm- rung, Wachstumshormon, GH, IGF-1 growth disorders was focused on whether the
varianten des Wachstums von nach heutigem child was growth hormone sufficient or insuffi-
Wissensstand behandelbaren und nichtbehandel- cient, based on the virtually unlimited availabili-
baren Wachstumsstörungen erschwert. Wichtige Abstract: Short Stature – Differential Diag- ty of recombinant growth hormone to treat these
Voraussetzung für eine Diagnose stellen die Erfas- nosis and Therapeutic Options. Assessment children. Since recombinant IGF-1 was commer-
sung der Parameter bei Geburt zur Erkennung ei- of children with growth disorders is of utmost cially launched a few years ago, new thera-
ner SGA-Konstellation, das Wissen über die Kör- importance for paediatricians in their routine peutic options evolved arguing for a revisited –
pergröße der biologischen Eltern, die Dokumenta- practice. Diagnosis is challenging because of the probably comparably artificial – classification of
tion des Wachstumsverlaufes eines Kindes durch wide spectrum of normal growth patterns and growth disorders. J Klin Endokrinol Stoffw
Führen einer geschlechts- und bevölkerungsspezi- maturation. Profound knowledge of physiological 2015; 8 (1): 5–14.
fischen Perzentilenkurve und das Erkennen einer growth is an important prerequisite to allow cor-
Disproportion, ebenfalls bestätigt durch Eintra- rect diagnosis. Careful collection of family histo- Key words: short stature, growth disorder,
gung der Sitzhöhen/Beinlängen-Ratio in eine Per- ry, birth weight and height, and precise analysis growth hormone, GH, IGF-1
Liste der verwendeten Abkürzungen
ACH Achondroplasie NVSS Normal-Variant Short Stature
ALS Acid-Labile Subunit PIGFD Primärer IGF-1-Mangel
CDGP Constitutional Delay of Growth and Puberty rhGH Rekombinantes humanes Wachstumshormon
EMA European Medicines Agency SD Standard Deviation
GH Wachstumshormon SDS Standard Deviation Score
HCH Hypochondroplasie SGA Small for Gestational Age
HSDS Height Standard Deviation Score SHOX Short Stature Homeobox-Containing Gene
IGF-1 Insulin-Like Growth Factor 1 SIGFD Sekundärer IGF-1-Mangel
IGF-2 Insulin-Like Growth Factor 2 SPIGFD Schwerer primärer IGF-1-Mangel
IGF-BP-3 Insulin-Like Growth Factor Binding Protein UTS Ullrich-Turner-Syndrom
IUGR Intrauterine Growth Restriction WH-R Wachstumshormonrezeptor
MPH Mid-Parental Height
Einleitung Kleinwuchs per se stellt noch keine Krankheitsentität dar und
muss von Wachstumsstörungen, die durch eine pathologische
Auffälligkeiten des Wachstums eines Kindes gehören zu den Wachstumsgeschwindigkeit unabhängig von der Körpergrö-
häufigsten Fragestellungen in der Pädiatrie. Zahlreiche Fakto- ße eines Kindes charakterisiert sind, als pathologische Wachs-
ren beeinflussen das Wachstum von Kindern: Hauptsächlich tumsform abgegrenzt werden. Die Differenzialdiagnose eines
verantwortlich sind genetische Faktoren, darüber hinaus spie- pathologischen Wachstums umfasst zunächst primäre/ossäre
len neben einer ausgewogenen Ernährung auch chronische Er- Wachstumsstörungen, d. h. dass die zugrunde liegende Patho-
krankungen, das soziokulturelle Umfeld und psychische Fak- logie im Skelettsystem selbst liegt. Diese kann entweder auf
toren eine wichtige Rolle (Abb. 1). der Basis von genetischen Erkrankungen (Knochenstoffwech-
selstörungen, Skelettdysplasien, metabolische Erkrankungen,
verschiedene Syndrome, Chromosomenaberrationen etc.) oder
Eingelangt am 23. Juni 2014; angenommen nach Revision am 10. August 2014 vorgeburtlichen (toxischen) Schädigungen (Nikotin, Alkohol,
Aus dem Department für Kinder- und Jugendheilkunde, Medizinische Universität Medikamente etc.) entstehen. Klinisch charakterisiert sind vie-
Innsbruck
le dieser Störungen durch eine mehr oder weniger ausgeprägte
Korrespondenzadresse: Dr. Klaus Kapelari, Leiter der Endokrinologischen Ambu-
lanz, Pädiatrie 1, Department für Kinder- und Jugendheilkunde, Medizinische Univer-
Disproportion, typischerweise mit zu kurzen Extremitäten im
sität Innsbruck, A-6020 Innsbruck, Anichstraße 35; Vergleich zum Oberkörper. Bei den sekundären Wachstums-
E-Mail: klaus.kapelari@i-med.ac.at störungen liegt die Ursache außerhalb des Skelettsystems, d. h.
J KLIN ENDOKRINOL STOFFW 2015; 8 (1) 5
For personal use only. Not to be reproduced without permission of Krause & Pachernegg GmbH.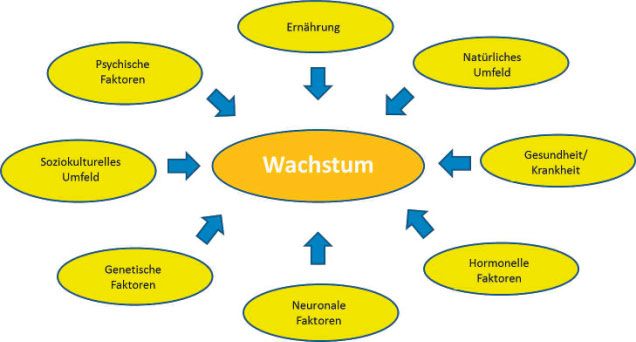
Kleinwuchs – Differenzialdiagnose und therapeutische Optionen
entweder auf hormoneller Ebene (GH, Schilddrüsenhormone) den auf, wobei die höchsten Spiegel während der Nacht beob-
oder ist verursacht durch chronische Erkrankungen (intestinal, achtet werden [3]. Die genaue Bedeutung der pulsatilen Sekre-
renal, hepatisch etc.), Störungen des Herz-Kreislauf-Systems, tion ist bis dato nicht endgültig geklärt. Eine wichtige Erkennt-
Mangelernährung oder Stoffwechselerkrankungen. Kinder mit nis aus allen Studien zu dieser Thematik ist jedoch auch, dass
sekundären Wachstumsstörungen weisen zumeist einen pro- mit den uns heute zur Verfügung stehenden Methoden eine klar
portionierten Kleinwuchs auf. Bei etwa jedem 4. kleinwüchsi- definierte Unterscheidung zwischen normaler und pathologi-
gen Kind kann nach heutigem Wissensstand eine Wachstums- scher GH-Sekretion nicht ausreichend möglich ist.
pathologie diagnostiziert werden.
Physiologisches Wachstum
Während der letzten 40 Jahre lag der Fokus der wissenschaft-
lichen und klinischen Arbeiten zum Thema Diagnose und Be- Intrauterin wird das Wachstum eines Kindes im Wesentlichen
handlung von Wachstumsstörungen auf dem Wachstumshor- durch das intrauterine Milieu und durch genetische sowie de-
mon (GH). Daran orientierte sich auch die Klassifikation der mographische Faktoren und Hormone gesteuert [4]. Die In-
Kleinwuchsformen mit den Hauptkriterien „mit Wachstums- sulin-ähnlichen Wachstumsfaktoren IGF-1 und IGF-2 neh-
hormonmangel“ und „ohne Wachstumshormonmangel“. Be- men für das Längenwachstum eine entscheidende Stellung
stimmt wurde diese Sicht durch die klinisch-diagnostischen ein: Während IGF-2 in der embryonalen Entwicklung die be-
Möglichkeiten der Durchführung von GH-Stimulationstests deutendere Rolle zugeordnet wird, erlangt gegen Ende der
und der Verfügbarkeit von rekombinantem Wachstumshor- Schwangerschaft und im weiteren Verlauf der somatischen
mon zur Behandlung von kleinwüchsigen Kindern. Die zuneh- Entwicklung IGF-1 zunehmend an Bedeutung [5, 6]. Ca. 75 %
mende Erkenntnis, dass IGF-1 und nicht GH die Schlüsselrol- des zirkulierenden IGF-1 wird in der Leber gebildet und in ei-
le in der Steuerung des Skelettwachstums einnimmt, und die nem ternären Komplex, bestehend aus IGF-1, IGF-BP-3 und
seit 2007 bestehende Zulassung der EMA (European Medici- der ALS („acid-labile subunit“), zur Verlängerung der Halb-
nes Agency) von rekombinantem IGF-1 für die Therapie des wertszeit gebunden. Die Regulation der hepatischen IGF-
Kleinwuchses im Rahmen eines „Orphan-drug“-Verfahrens er- 1-Synthese und auch die Regulation der IGF-1-Sensitivität
fordern nun ein Umdenken. Wenn auch möglicherweise ähn- in der Epiphysenfuge erfolgen gemäß traditioneller Hypothe-
lich arbiträr wie die GH-zentrierte Klassifikation, so eröffnet sen im Wesentlichen über Wachstumshormon. Der exakte Me-
eine Unterscheidung in „Wachstumsstörungen mit primärem chanismus der lokalen IGF-1-Produktion und der Regulation
IGF-1-Mangel“ (vermindertes IGF-1 bei normalen/erhöhten durch GH ist jedoch noch nicht völlig geklärt [7, 8].
GH-Spiegeln [PIGFD]) und „Wachstumsstörungen mit sekun-
därem IGF-1-Mangel“ (vermindertes IGF-1 und verminder- Als normales Geburtsgewicht bzw. normale Geburtslänge wer-
te GH-Spiegel [SIGFD]) die Möglichkeit eines differenzierte- den Werte innerhalb von ± 2 Standardabweichungen (SD) be-
ren, physiologischeren therapeutischen Ansatzes. Neben IGF- zeichnet. Die für Österreich von der Arbeitsgruppe für Pädia-
1 und IGF-BP-3 werden in der Diagnostik zusätzlich die basa- trische Endokrinologie und Diabetologie (APEDÖ) empfohle-
len sowie die stimulierten GH-Werte, der IGF-Generationstest nen Referenzwerte wurden im Jahr 2008 in der Monatsschrift
und die Erstellung von GH-Sekretionsprofilen eingesetzt [1]. Kinderheilkunde von Mayer et al. publiziert [9]. Bei Termin-
Im Hinblick auf die Diagnosestellung einer PIGFD ist die Er- geborenen wird diesen Daten zufolge als Normalgewicht (± 2
mittlung der normalen GH-Sekretion eine wichtige Vorausset- SD) für einen Knaben ein Bereich zwischen 2709 und 4403 g,
zung zur Unterscheidung zwischen pathologischen und phy- für Mädchen zwischen 2664 und 4286 g angeführt. Die nor-
siologischen GH-Sekretionsraten [2]. Die Bestimmung von male Geburtslänge (± 2 SD) liegt für termingeborene Knaben
GH-Sekretionsprofilen zeigt, dass neben der zirkadianen auch zwischen 47,5 und 56,1 cm und für Mädchen zwischen 47 und
die infradiane und ultradiane Rhythmik der GH-Sekretion für 55 cm. In den ersten 1–3 (–4) Lebensjahren erreicht ein Kind
ein normales Wachstum von Bedeutung sind [3]. Die Sekre- dann jenen Perzentilenbereich in der Normalverteilungskurve,
tionsprofile gesunder Kinder weisen ca. 8 GH-Pulse in 24 Stun- der durch den genetischen Zielbereich vorgegeben wird. Das
bedeutet, dass ein Kreuzen der Perzenti-
len in den ersten Lebensjahren nicht mit
dem Vorliegen einer Wachstumsstörung
gleichgesetzt werden kann. Der mittle-
re genetische Zielbereich („mid-paren-
tal height“ [MPH]) errechnet sich aus
der Körpergröße der Eltern eines Kin-
des. Das „normale“ Wachstum folgt an-
schließend weitgehend dieser Perzenti-
le, wobei physiologischerweise die jähr-
liche Wachstumsrate kontinuierlich ab-
nimmt. Die letzte Phase des Wachstums
wird durch die Pubertätsentwicklung
und somit entscheidend durch die Wir-
kung der Sexualhormone auf die GH-
Sekretion und die Wachstumsfuge be-
stimmt [10]. Auch in dieser sehr indivi-
Abbildung 1: Einflüsse auf das normale Wachstum von Kindern duellen Phase des Wachstums kann es zu
6 J KLIN ENDOKRINOL STOFFW 2015; 8 (1)
Kleinwuchs – Differenzialdiagnose und therapeutische Optionen
Alter ш 3 Jahre nein Größe < –3 SDS nein nein
2× Größe < –2,5 SDS Keine Abklärung
ja
ja
ja
ja
Größe < –2,5 SDS Zuweisung Pädiatrische Endokrinologie
nein
ja
ja
Größe < –2,0 SDS nein Anamnese SGA nein ŝƐƉƌŽƉŽƌƟŽŶ
Dysmorphie
ja
nein
nein
ja
Keine Abklärung Größe < genet. ZB
HSDS – TSDS < –2 SDS
nein
nein
Abfall der Wachstumsgeschw.
< –1 SDS
Abbildung 2: Evidenzbasierte Kriterien für eine Zuweisung an eine spezialisierte Ambulanz für pädiatrische Endokrinologie zur Wachstumsabklärung. Mod. nach [59].
SGA: „small for gestational age“; SDS: „standard deviation score“; HSDS: „height standard deviation score“; TSDS: „target height standard deviation score“; ZB: Zielbereich
einem Kreuzen der Perzentile kommen, wenn auch letztend- tem Kleinwuchs müssen im nächsten Schritt organische Ur-
lich der genetisch determinierte Perzentilenbereich beibehal- sachen des Kleinwuchses (z. B. Zöliakie, chronisch entzünd-
ten wird. liche Erkrankungen, Vitien, Niereninsuffizienz, Leberfunk-
tionsstörungen etc.) und eine psychosoziale Deprivation aus-
In einer Querschnittsstudie der Arbeitsgruppe für Pädiatrische geschlossen werden. Erst auf dieser Stufe der Abklärung
Endokrinologie und Diabetologie (APEDÖ) der Österreichi- erfolgt die traditionell zur Berechnung der Endlängenprogno-
schen Gesellschaft für Kinder- und Jugendheilkunde wurden se eines Kindes eingesetzte Knochenalterbestimmung aus ei-
im Zeitraum zwischen 2009 und 2011 ca. 14.500 Kinder an nem Skelettröntgen der linken Hand und die Bestimmung der
österreichischen Kindergärten und Schulen von einem erfah- Hormonwerte für IGF- 1 und IGF-BP-3 [12–14]. Die Korrela-
renen pädiatrischen Endokrinologen vermessen und daraus tion des chronologischen Alters mit dem Knochenalter ist ein
repräsentative Referenzwerte für die österreichische Bevölke- wichtiger differenzialdiagnostischer Parameter.
rung erstellt [11].
Ein erheblicher Anteil der Kinder, die zur Wachstumsabklä-
Proportionierter Kleinwuchs rung vorgestellt werden, hat ebenfalls kleine Eltern. Das gene-
tische Wachstumspotenzial dieser Kinder kann durch die Be-
Normvarianten des Wachstums rechnung der genetischen Zielgröße abgeschätzt werden. Es
Kleinwuchs ist definiert als Körpergröße eines Kindes unter- existiert hierzu eine Vielzahl an Berechnungsmethoden. In der
halb der 3. Perzentile (bezogen auf Geschlecht, Alter und Po- Praxis hat sich jedoch durchgesetzt, die mittlere Größe bei-
pulation). Alternativ kann auch die standardisierte Abwei- der Eltern zu verwenden (möglichst gemessen) und bei Kna-
chung vom Mittelwert der Referenzpopulation („standard de- ben 6,5 cm zu addieren, bei Mädchen zu subtrahieren. Im Be-
viation score“ [SDS]) herangezogen werden, wobei die 3. Per- reich von ± 8,5 cm zur genetischen Zielgröße liegt der sog.
zentile in etwa einem Wert von –2,3 SD entspricht. Kleinwuchs genetische Zielbereich [15]. Bei der Berechnung des geneti-
kann bereits bei Geburt bestehen (primärer Kleinwuchs) oder schen Zielbereiches muss jedoch die Möglichkeit berücksich-
im weiteren Verlauf durch zu geringes oder zu früh endendes tigt werden, dass bereits ein Elternteil eine in früherer Zeit
Längenwachstum entstehen (sekundärer Kleinwuchs). Sei- nicht diagnostizierte Wachstumsstörung aufweisen könnte,
tens der pädiatrisch-endokrinologischen Fachgesellschaften die zu einer Verfälschung der Berechnung führen würde. In
wurden evidenzbasierte Kriterien für eine Zuweisung an eine den ersten 2–4 Jahren weisen Kinder mit familiärem Klein-
Wachstumsambulanz entwickelt (Abb. 2). wuchs typischerweise eine geringe Wachstumsgeschwindig-
keit auf und kreuzen daher nicht selten, ausgehend von einer
In einem ersten Schritt müssen die auxologischen Parame- normalen Geburtsgröße, die Perzentilen nach unten, bis sie
ter eines Kindes inkl. der Körperproportionen ermittelt und in den genetisch vorgegebenen Perzentilenbereich erreicht ha-
die jeweiligen Perzentilenkurve übertragen werden, um einen ben. Bestimmt man die Werte für IGF-1 und IGF-BP-3, so lie-
proportionierten von einem disproportionierten Kleinwuchs gen diese im Normalbereich. Das Knochenalter dieser Kin-
abzugrenzen [11]. Ein disproportionierter Kleinwuchs weist der entspricht im Wesentlichen dem chronologischen Alter,
auf eine ossäre/genetische Ursache hin. Bei proportionier- d. h. liegt im Bereich von ± 1 Jahr mit dem chronologischen
J KLIN ENDOKRINOL STOFFW 2015; 8 (1) 7
Kleinwuchs – Differenzialdiagnose und therapeutische Optionen
Alter. Einige der Kinder, die zur Wachstumsabklärung vorge- schiedenen Endokrinopathien (z. B. Wachstumshormonman-
stellt werden, weisen ebenfalls normale Werte für IGF-1 und gel, Hypothyreose etc.) auf. Kinder mit schwerem primärem
IGF-BP-3, ein normales Knochenalter und jenseits des 4. Le- IGF- 1-Mangel (SPIGFD) weisen einen ausgeprägten Klein-
bensjahres eine normale Wachstumsgeschwindigkeit auf, je- wuchs (Körpergröße < 3,0 SDS) auf, ausgenommen jene mit
doch wachsen sie unterhalb ihres genetischen Zielbereiches. einem Mangel der säurelabilen Untereinheit (ALS), deren
Diese Kleinwuchsform wird als „idiopathischer Kleinwuchs“ Wachstum nur wenig bis gar nicht beeinträchtigt ist [19]. Die
bezeichnet, wobei eine primäre Wachstumsstörung ausge- basalen Konzentrationen von IGF-1 (und IGF-BP-3) liegen
schlossen werden muss. bei der SPIGFD unter der 2,5-ten Perzentile (bezogen auf Al-
ter und Geschlecht). In Wachstumshormonstimulationstests
Neben dem familiären und dem idiopathischen Kleinwuchs weisen diese Kinder einen normalen bis überschießenden An-
stellt die sog. konstitionelle Verzögerung von Wachstum stieg des Wachstumshormons auf, sie sind also Wachstums-
und Pubertät („constitutional delay of growth and puberty“ hormon-suffizient. Ein IGF-1-Generationstest kann helfen,
[CDGP]) eine weitere Normvariante des Wachstums dar, wo- die Diagnose biochemisch zu bestätigen: Dabei wird rekom-
bei Knaben deutlich häufiger betroffen sind als Mädchen. De- binantes Wachstumshormon an 7 Tagen in einer täglichen Do-
finitionsgemäß kann diese Kleinwuchsform erst im Alter des sis von 35 µg/kg s.c. injiziert und der Anstieg des IGF-1 ge-
physiologischen Pubertätseintritts, also im Alter von 13 Jah- messen [1].
ren bei Mädchen und 14 Jahren bei Knaben, gestellt werden.
In der Kindheit sind jedoch ein Wachstum unterhalb der ge- Bereits im Jahr 1966 wurde von Laron et al. eine nach ihm be-
netischen Zielgröße, eine niedrig-normale Wachstumsge- nannte Kleinwuchsform mit suffizienter Wachstumshormon-
schwindigkeit, normale Werte für IGF-1 und IGF-BP-3 und sekretion, die phänotypisch die Merkmale eines Wachstums-
> 1 Jahr retardiertes Knochenalter charakteristisch. Die be- hormonmangels aufwies [20], beschrieben. Die von den Au-
troffenen Kinder weisen einen verspäteten spontanen Puber- toren als ursächlich postulierte aufgehobene Wirksamkeit von
tätseintritt auf; die Familienanamnese diesbezüglich ist häufig Wachstumshormon konnte fast 20 Jahre später mit der Wei-
positiv. Durch den verzögerten Pubertätseintritt kommt es im terentwicklung molekulargenetischer Methoden als Mutation
Alter des physiologischen Pubertätseintritts zu einem Kreu- des Wachstumsrezeptorgens bestätigt werden [21]. Die zu-
zen der Perzentile nach unten und es müssen daher angepasste nehmenden Erkenntnisse über die durch Bindung von Wachs-
Normwerte für die jährliche Wachstumsgeschwindigkeit der tumshormon an den Wachstumshormonrezeptor (WH-R) ver-
Beurteilung zugrunde gelegt werden [16]. Die CDGP stellt mittelte Signalkaskade zur Stimulation der IGF-1-Expression
somit eine zeitliche Variante eines normalen Wachstums dar; führte zur Identifikation von weiteren monogenetischen Ur-
die Endgröße der betroffenen Kinder ohne therapeutische In- sachen einer Wachstumshormonresistenz. Mutationen in den
tervention liegt innerhalb ihres genetischen Zielbereichs. Der kodierten Sequenzen des WH-R für STAT5b („signal trans-
Leidensdruck durch den Kleinwuchs wird jedoch besonders ducers and activators of transcription“), IGF-1 und ALS füh-
zum Zeitpunkt des physiologischen Pubertätseintritts evident. ren biochemisch zur typischen Konstellation einer Wachs-
Eine kurzzeitige 3-monatige Therapie mit niedrig dosierten tumshormonresistenz mit hohen Wachstumshormonspiegeln
Sexualsteroiden (Testosteronenantat 50 mg i.m. monatlich bei erniedrigtem IGF-1 [2, 22]. Liegen gleichzeitig erhöhte
bzw. Estradiolvalerat 0,2 mg p.o. täglich) kann in diesen Fäl- Nüchtern-Insulinkonzentrationen vor, kann die Bestimmung
len im Sinne eines Pubertätsprimings in Erwägung gezogen einer erniedrigten ALS im Serum helfen, die Diagnose einer
werden. SPIGFD zu sichern und schließlich molekulargenetisch zu be-
stätigen. Bei Mutationen des IGF-1-Gens, die zur Synthese
Die in diesem Abschnitt dargestellten Kleinwuchsformen von bioinaktivem IGF-1 führen, sind sowohl die Wachstums-
werden auch als „Normvarianten des Wachstums“ bezeichnet hormonspiegel als auch die Spiegel für IGF-1 erhöht.
und sind ca. 100× häufiger als Kleinwuchs durch Wachstums-
hormonmangel. Sie bedürfen keiner therapeutischen Interven- 1988 wurden erste Therapieversuche mit intravenöser Injek-
tion und stellen in Europa auch keine anerkannten Indikatio- tion von IGF-1 bei Patienten mit Wachstumshormonresistenz
nen zur Therapie mit rekombinantem Wachstumshormon dar. unternommen [23]. Das rasche Auftreten von Hypoglykämi-
In den USA hingegen wurde im Jahr 2003 die Indikationslis- en nach der Injektion wurde als Hinweis auf die Wirksamkeit
te für rekombinantes Wachstumshormon (rhGH) um die The- und die möglichen positiven Langzeiteffekte gewertet. Die in
rapie von Kindern mit „idiopathic short stature“ bzw. „nor- der Folge beobachteten Nebenwirkungen einer s.c. Therapie
mal-variant short stature“ (NVSS) erweitert. Erstmalig wur- mit rekombinantem IGF-1 beschränkten den therapeutischen
de somit rhGH neben den pathologischen (extremen) Klein- Einsatz weltweit auf die wenigen Patienten mit Laron-Klein-
wuchsformen offiziell zur Therapie eines nichtpathologischen wuchs, die auf eine Therapie mit rekombinantem Wachstums-
Kleinwuchses zugelassen. Eine diesbezügliche Zulassung in hormon nicht ansprachen. Erst die Aufklärung von weiteren
Europa ist bis dato nicht erfolgt und Gegenstand zahlreicher genetischen Defekten einer Wachstumshormonresistenz führ-
fachlicher Diskussionen [17]. te zu einer Erweiterung der möglichen Indikationen und dazu,
dass seit 2007 rekombinantes IGF-1 von der EMA für die
Primärer IGF-1-Mangel (PIGFD) Therapie der SPIGFD im Rahmen eines „Orphan-drug“-Ver-
Monogenetische Ursachen eines IGF-1-Mangels sind insge- fahrens zugelassen wurde. Rekombinantes IGF-1 muss 2× tgl.
samt sehr selten (Inzidenz < 1:20.000) [18]. Sehr viel häufi- s.c. injiziert und einschleichend dosiert werden. Die Initialdo-
ger tritt der IGF-1-Mangel als sekundäres Phänomen ande- sis beträgt 0,04 mg/kg/Dosis 2× tgl. und wird bei guter Ver-
rer Erkrankungen und hier besonders bei Malnutrition, aber träglichkeit im Verlauf mehrerer Wochen auf eine Dosis bis zu
auch bei Adipositas, chronischen Erkrankungen und ver- 0,12 mg/kg/Dosis 2x tgl. gesteigert. Bei jeder Dosissteigerung
8 J KLIN ENDOKRINOL STOFFW 2015; 8 (1)
Kleinwuchs – Differenzialdiagnose und therapeutische Optionen
muss auf das Auftreten von Hypoglykämien geachtet und die Die Diagnose eines Wachstumshormonmangels ist primär eine
IGF-1-Werte monitorisiert werden. klinische Diagnose, die sekundär durch laborbiochemische
Parameter untermauert wird. Neben der typischen Physiogno-
Der Erfolg einer Therapie mit rekombinantem IGF-1 (Meca- mie und dem proportionierten Kleinwuchs steht der Nachweis
sermin) bei Patienten mit SPIGFD im Hinblick auf die Ver- eines pathologischen Wachstums mit Perzentilenflucht nach
besserung der Endgröße ist geringer als jener einer Thera- unten im Zentrum der Diagnose. Eine weitere wichtige Säu-
pie mit rekombinantem Wachstumshormon bei Patienten mit le der Diagnostik ist die Bestimmung des Knochenalters aus
Wachstumshormonmangel. Es ist jedoch mit einer Verbesse- einem Handskelettröntgen links, das typischerweise eine Re-
rung der Endgröße um ca. +2 SDS zu rechnen, wenngleich tardierung von > 1 Jahr aufweist. Nur wenn diese klinischen,
die Patienten auch meist nur eine Erwachsenengröße unter- radiologischen und auxologischen Hinweise vorliegen, sol-
halb des Normalbereiches für die jeweilige Population errei- len die Werte für IGF-1 und IGF-BP-3 bestimmt werden, wo-
chen [24]. Aufgrund der enthaltenen Konservierungsmittel in bei diese zwei Laborparameter im engeren Sinne keine Scree-
Mecasermin ist eine Therapie bei Kindern < 2 Jahren nicht ningparameter darstellen, da sie auch bei verschiedenen ande-
zugelassen. Zudem stellen aktive onkologische Erkrankungen ren Erkrankungen vermindert sein können. Bestätigt wird das
eine strikte Kontraindikation für eine Therapie mit rekombi- Vorliegen eines Wachstumshormonmangels durch die Durch-
nantem IGF-1 dar. Die Therapie sollte nur von einem erfah- führung von zwei Wachstumshormonstimulationstests. Zur
renen pädiatrischen Endokrinologen durchgeführt und mög- Austestung stehen Insulin, Glukagon, Arginin oder Clonidin
lichst alle behandelten Patienten in Postmarketing-Datenban- zur Verfügung. Vorsicht ist geboten mit den Testsubstanzen
ken erfasst werden. Glukagon und Insulin bei Kindern < 4 Jahren, da schwere und
bei Insulin zudem bedrohliche Hypokaliämien auftreten kön-
Sekundärer IGF-1-Mangel (SIGFD) nen [26]. Sensitivität und Spezifität der Stimulationstests lie-
Viel häufiger als durch eine PIGFD wird Kleinwuchs durch gen mit dem aktuell angewendeten Grenzwert der maxima-
einen sekundären IGF-1-Mangel (SIGFD) verursacht. Die zu- len GH-Ausschüttung von 8 µg/l bei ca. 80 %. Aussagekräftig
grunde liegenden Ursachen sind vielfältig: Am häufigsten füh- sind die Tests somit nur, wenn auch die klinischen, radiologi-
ren Mangelernährung, aber auch Adipositas sowie chronische schen und auxologischen Kriterien bei gleichzeitig erniedrig-
Erkrankungen und verschiedene Endokrinopathien (z. B. Hy- ten Werten für IGF-1 und IGF-BP-3 erfüllt sind. Knaben ab
pothyreose) zu einer Erniedrigung des zirkulierenden IGF-1. einem Alter von 10 Jahren und Mädchen ab einem Alter von 8
Die Therapie dieser Kleinwuchsformen erfolgt durch die Be- Jahren müssen zudem vor Testdurchführung ein Priming mit
handlung der zugrunde liegenden Störung. Sexualsteroiden erhalten, da die Wachstumshormonsekretion
physiologisch in der präpubertären Wachstumsphase verrin-
Die häufigste endokrine Ursache einer SIGFD und insgesamt gert ist. Mädchen erhalten hierzu 1 mg Estradiolvalerat p.o. an
die häufigste endokrine Ursache eines pathologischen Wachs- 3 Tagen vor dem Test, Knaben entweder 50 mg Testosteronen-
tums ist der Wachstumshormonmangel. Die Prävalenz wird antat i.m. 7 Tage vor dem Test oder alternativ 40 mg Testoste-
auf 1:4000–1:30.000 geschätzt. Bei der überwiegenden An- ronundecanoat p.o. an 5 Tagen vor dem Test [27, 28].
zahl von Kindern wird ein idiopathischer Wachstumshormon-
mangel diagnostiziert. Daneben werden selten auch mono- Ein normaler Anstieg des Wachstumshormons im Stimula-
genetische Ursachen gefunden, die autosomal-rezessiv oder tionstest schließt einen Wachstumshormonmangel praktisch
-dominant vererbt werden und zu isolierten (GH-1) oder aus. Es muss eine Neuevaluierung auf das Vorliegen anderer
kombinierten (PIT-1, PROP-1, HESX-1, LHX4, SOX3) hy- Ursachen einer Wachstumsstörung erfolgen. Bei persistierend
pophysären Hormonausfällen führen. Letztere betreffen kriti- pathologischer Wachstumsgeschwindigkeit ohne diagnosti-
sche Transkriptionsfaktoren der Organogenese von Hypotha- zierbare Ursache kann der Stimulationstest nach einem Jahr
lamus und Hypophyse und führen zu morphologischen Verän- wiederholt werden. Bei normalem, jedoch in einigen Fällen
derungen der Hypophyse, teilweise in Kombination mit kom- zeitlich verzögertem Anstieg des Wachstumshormons und
plexeren Fehlbildungen des Gehirns. Darüber hinaus können fortbestehendem pathologischem Wachstum können die Mes-
isolierte und kombinierte hypophysäre Hormonausfälle auch sung des nächtlichen rhGH-Sekretionsprofils mit Nachweis
nach zerebralen Infektionen, durch Trauma, infiltrative Er- einer verminderten Amplitude und die Anzahl von Wachs-
krankungen oder iatrogen nach Bestrahlung oder neurochirur- tumshormon-Peaks während der Nachtstunden dem Beleg ei-
gischen Eingriffen auftreten. ner neurosekretorischen Dysfunktion dienen [29]. Ein über-
schießender Anstieg der Wachstumshormonausschüttung
Kinder mit angeborenem Wachstumshormonmangel werden ty- in Kombination mit erniedrigten Werten für IGF-1und IGF-
pischerweise mit normaler Geburtslänge geboren, jedoch meist BP-3 spricht für das Vorliegen einer Wachstumshormonresis-
bereits in der Neugeborenenperiode durch rezidivierende Hy- tenz, also einer SPIGFD, die durch einen IGF-1-Generations-
poglykämien symptomatisch. Zusätzlich kann bei Knaben ein test gesichert werden kann [1]. Bei positivem Nachweis einer
Mikropenis auf das Vorliegen einer kombinierten hypophysären verminderten Wachstumshormonsekretion muss eine zerebra-
Insuffizienz hinweisen. Die Bestimmung des Wachstumshor- le Magnetresonanztomographie mit Kontrastmittel zum Aus-
mons aus der Trockenblutkarte des Neugeborenenscreenings schluss einer Raumforderung im Bereich der Hypophyse, am
innerhalb der ersten Lebenswoche kann die Diagnose sichern häufigsten eines Kraniopharyngeoms, als Ursache des Wachs-
[25]. Der umgehende Beginn einer Therapie mit rekombinan- tumshormonmangels durchgeführt werden. Die Bildgebung
tem Wachstumshormon ist bei diesen Kindern bereits in der dient zudem der Darstellung von charakteristischen Fehlbil-
Neugeborenenperiode aufgrund der komplexen metabolischen dungen der Hypophyse bzw. komplexen Fehlbildungen des
Konsequenzen der Wachstumshormondefizienz unerlässlich. Gehirns.
J KLIN ENDOKRINOL STOFFW 2015; 8 (1) 9
Kleinwuchs – Differenzialdiagnose und therapeutische Optionen
1957 wurde Kleinwuchs erstmals erfolgreich mit humanem pie regelmäßig zu messen und im Normalbereich für das je-
Wachstumshormon behandelt. Der Einsatz von humanem weilige Alter zu halten.
Wachstumshormon war jedoch aufgrund der beschränkten
Verfügbarkeit auf die Behandlung des Wachstumshormon- Intrauteriner Kleinwuchs – SGA
mangels limitiert. 1985 tauchten bei internationalen Tagun- Eine sich bereits intrauterin manifestierende Wachstumsstö-
gen erste Berichte über Creutzfeldt-Jakob-Erkrankungen bei rung wird als „intrauterine growth restriction“ (IUGR) be-
Patienten, die mit humanem Wachstumshormon behandelt zeichnet. Als Folge der intrauterinen Wachstumsstörung wer-
wurden, auf [30]. Nahezu zeitgleich mit dem Auftreten die- den die Kinder zu klein und/oder zu leicht geboren (bezo-
ser Berichte wurde rekombinantes humanes Wachstumshor- gen auf die bevölkerungsspezifischen Normalverteilungskur-
mon (rhGH) zur Behandlung von kleinwüchsigen Kindern ven für das jeweilige Gestationsalter und das Geschlecht) [9].
mit Wachstumshormonmangel eingeführt. Die Therapie mit Diese Kinder werden als SGA-Kinder („small for gestational
rhGH erfolgt durch tägliche abendliche subkutane Injektio- age“) bezeichnet. Die Begriffe IUGR und SGA werden häufig
nen in einer Dosis von 25–35 µg/kg unter Verwendung ei- synonym verwendet, obwohl nicht obligat jeder SGA-Konstel-
nes Pen-Systems. In den ersten 1–2 Jahren der Therapie ver- lation bei Geburt eine IUGR, die definitionsgemäß einen oder
zeichnen Kinder mit Wachstumshormonmangel einen be- mehrere intrauterin wirksame wachstumshemmende Einflüs-
achtlichen Anstieg der Wachstumshormontherapie auf 8–12 se impliziert (dokumentiert durch 2 oder mehrere intrauterine
cm/Jahr. Das Ansprechen auf die Therapie in den ersten 1–2 Vermessungen, die eine verminderte Wachstumsgeschwindig-
Behandlungsjahren dient auch als wichtiger Indikator für den keit belegen), zugrunde liegen muss. Daneben werden auch
gesamten Erfolg der Therapie [31]. Nach Abschluss des Auf- Kinder, die bereits zum Zeitpunkt der Geburt charakteristische
holwachstums sinkt die jährliche Wachstumsgeschwindig- Auffälligkeiten aufweisen, wie z. B. das Russel-Silver-Syn-
keit unter der Therapie in den Normalbereich ab und behan- drom, in dem Begriff „SGA“ erfasst. Die Heterogenität der
delte Kinder wachsen vergleichbar zu ihren Altersgenossen. verschiedenen Entitäten, die unter dem Begriff „SGA“ zusam-
Gelingt es unter der Therapie, das Aufholwachstum bis zum mengefasst werden, erschwert somit auch die Erstellung all-
Eintreten der spontanen Pubertätsentwicklung abzuschlie- gemeingültiger Aussagen zu SGA-Kindern. „SGA“ ist somit
ßen, erreichen behandelte Kinder eine Endgröße im Bereich ein rein deskriptiver Begriff und keine eigenständige Entität.
ihrer genetischen Zielgröße. Der optimale Zeitpunkt für den
Therapiebeginn bei Kindern mit hypophysärem Kleinwuchs Etwa 5 % der reifen neugeborenen Kinder sind bei Geburt zu
liegt in etwa im 5. Lebensjahr, jedoch ist er bei annähern- klein und/oder zu leicht (bezogen auf das Gestationsalter). Bei
dem Wachstumsstillstand oder bei Kindern mit angebore- Frühgeborenen ist dieser Prozentsatz deutlich höher und liegt
nem Wachstumshormonmangel bereits früher zu empfehlen. bei ca. 30 %. Die zu geringen Geburtsmaße sind einerseits
Ein Behandlungsbeginn in der präpubertären Phase oder bei durch mütterliche und fetale genetische Faktoren, aber auch
bereits eingetretener Pubertät ist mit deutlich geringeren Er- durch das intrauterine Milieu bedingt. Anfang der 1970er-Jah-
folgsaussichten verbunden und daher häufig nicht mehr zu re prägte G. Dörner den Begriff der „intrauterinen Program-
empfehlen. Die Therapie wird bis zum fast vollständigen Ver- mierung“ [38]. Diesem Ansatz liegt das Konzept zugrunde,
schluss der Wachstumsfugen (Knochenalter bei Knaben ca. dass durch ein alteriertes Intrauterinmilieu Hormone wie In-
16–17 Jahre, bei Mädchen ca. 15–16 Jahre) bzw. bis zum Ab- sulin, Leptin, Kortisol und Neurotransmitter wie Neuropeptid
sinken der jährlichen Wachstumsgeschwindigkeit auf einen Y, Galanin in anormalen Konzentrationen vorliegend als sog.
Wert < 2 cm/Jahr fortgeführt. Nach Beendigung der Therapie funktionelle Teratogene, die zu einer lebenslangen Fehlorga-
sollte bei Patienten mit idiopathischem Wachstumshormon- nisation von Regelkreisen und somit erhöhter Krankheitsprä-
mangel in angemessenem Intervall die Wachstumshormon- disposition führen, fungieren. Spätere Studien schrieben be-
suffizienz kontrolliert werden und ggf. nach neuerlicher Aus- sonders auch dem Übergang von einer fetalen Unterernährung
testung im Falle einer unzureichenden Wachstumshormon- zu einer frühpostpartalen Überernährung eine entscheidende
sekretion eine lebenslange Therapie mit Wachstumshormon Rolle in der lebenslangen Störung metabolischer und neuro-
erfolgen. Eine Therapieunterbrechung bzw. eine neuerliche endokriner Regelkreise zu (Abb. 3) [39].
Austestung ist bei genetisch verifiziertem schwerem Wachs-
tumshormonmangel bzw. bei iatrogenem Hypopituitarismus Der überwiegende Teil der SGA-Kinder (ca. 85–90 %) zeigt
nicht erforderlich. In diesen Fällen erfolgt eine fließende Do- ein spontanes Aufholwachstum innerhalb von 6 Monaten, Ter-
sisanpassung an die empfohlene Dosierung von rhGH im Er- mingeborene längstens innerhalb von 2 Jahren, Frühgeborene
wachsenenalter. etwas verzögert innerhalb von 3–4 Jahren. Wie in großen re-
trospektiven Studien gezeigt werden konnte, waren ungefähr
Mögliche Nebenwirkungen der rhGH-Therapie (z. B. Pseudo- 20 % der kleinwüchsigen Erwachsenen SGA-Kinder.
tumor cerebri, Epiphysiolysis capitis femoris oder Diabe-
tes mellitus Typ 2) treten dosisabhängig auf [32, 33]. Rezen- Die Stimulation des Aufholwachstums und die Verbesserung
te Publikationen einer erhöhten Langzeitmortalität nach ei- der Endlänge bei kleinwüchsigen präpubertären SGA-Kindern
ner rhGH-Therapie zeigen ebenfalls eine Dosisabhängigkeit mit supraphysiologischen Dosierungen von rhGH wurden in
und konnten in anderen Beobachtungen nicht bestätigt wer- verschiedenen nationalen und internationalen Studien belegt
den [34–37]. Die EMA erklärte daher ausdrücklich ein posi- [40]. Im Sommer 2001 erfolgte daher in den USA die Zulas-
tives Risiko-Nutzen-Profil der rhGH-Therapie und empfiehlt, sung von rhGH für die Langzeitbehandlung von Wachstums-
die empfohlenen Höchstdosen einzuhalten und nicht > 50 µg/ störungen bei SGA-Kindern durch die Food and Drug Admi-
kg/d zu therapieren. Unter diesen Aspekten ist es nach heuti- nistration (FDA), im Juli 2003 durch die EMA auch in Europa
gem Wissensstand notwendig, die IGF-1-Werte unter Thera- (Indikationskriterien siehe Tab. 1). In einer holländischen Stu-
10 J KLIN ENDOKRINOL STOFFW 2015; 8 (1)
Kleinwuchs – Differenzialdiagnose und therapeutische Optionen
Tabelle 1: Von der EMA (European Medicines Agency) Fetale und/oder frühpostnatale Intrauterine Wachstumsverzögerung –
Überernährung niedriges Geburtsgewicht
anerkannte Indikationskriterien einer Therapie mit rekombi-
nantem Wachstumshormon bei SGA-Kindern
Geburtsgewicht und/oder < –2 SD
Geburtslänge Perinataler Hyperinsulinismus
Hyperleptinismus
Aktuelle Körpergröße < –2,5 SDS Hyperkortisolismus
Wachstumsgeschwindigkeit < 0 SDS
Eltern-angepasste Zielhöhe < –1 SDS
Alter des Kindes 4 Jahre
SDS: „standard deviation score“
die an SGA-Kindern unter Langzeittherapie mit rhGH konnte
gezeigt werden, dass 85 % der Kinder eine Erwachsenengrö- Perinatal erworbene Disposition für
Adipositas, Diabetes mellitus und
ße im Normalbereich und 98 % eine Endgröße innerhalb ih- metabolisches Syndrom
res genetischen Zielbereichs erreichen [41]. Die Heterogenität
der verschiedenen Entitäten unter dem Begriff „SGA“ bedingt Abbildung 3: Konzept des „small baby syndrome“. Nicht die fetale Unterernährung
und das niedrige Geburtsgewicht, sondern der Übergang von fetaler Unterernährung
jedoch ein sehr individuelles Ansprechen auf die Therapie. auf frühpostnatale Überernährung führen zur Prädisposition für Adipositas, zu erhöhtem
artherogenen Risiko und schließlich zur Entwicklung eines metabolischen Syndroms.
Neben dem positiven Effekt auf die Körpergröße verlagerte
sich besonders in jüngster Zeit das Interesse zahlreicher Ar- und frühpostnatalen epigenetischen Modifikation von meta-
beitsgruppen auf die weiteren, vor allem metabolischen Aus- bolischen und neuroendokrinen Regelkreisen dient als Basis
wirkungen der Therapie mit rhGH. Besonders die Zusam- der aktuellen wissenschaftlichen Untersuchungen der meta-
menhänge zwischen niedrigem Geburtsgewicht und stattfin- bolischen Störungen bei SGA-Kindern mit dem Ziel der Ent-
dendem bzw. fehlendem postpartalem „catch-up“ und der wicklung präventiver Strategien.
späteren Entwicklung einer Adipositas und Insulinresistenz,
einhergehend mit Diabetes mellitus Typ 2, einem metaboli- Disproportionierter Kleinwuchs
schen Syndrom und assoziierten kardiovaskulären Komplika-
tionen, stehen dabei im Zentrum des Interesses. Ullrich-Turner-Syndrom
Mit einer Inzidenz von ca. 1:2000 weiblichen Neugeborenen
Der Zusammenhang zwischen niedrigem Geburtsgewicht stellt das Ullrich-Turner-Syndrom (UTS) die häufigste syndro-
und gestörter Glukosehomöostase wurde bereits Anfang der male Kleinwuchsform bei Mädchen dar. Dem Syndrom liegt
1990er-Jahre beschrieben. Pathophysiologisch wurde primär eine numerische oder strukturelle Aberration des X- Chromo-
von einer gestörten Insulinsekretion auf Basis einer gestörten soms zugrunde, die zu der typischen Trias aus Kleinwuchs,
Betazellfunktion, verursacht durch einen intrauterinen Nähr- Gonadendysgenesie mit unterschiedlich ausgeprägter ovariel-
stoffmangel, ausgegangen [42, 43]. Spätere Studien, die eine ler Insuffizienz und Minoranomalien (u. a. Nageldysplasien,
normale Anzahl und Dichte der Pankreasinseln sowie eine inverser Haaransatz, Pterygium colli, Ptosis, positives Meta-
normale Morphologie der Betazellen zeigten, belegen, dass carpalzeichen, Cubitus valgus und Linksherzfehlbildungen) in
eine Resistenz verschiedener Zielorgane gegenüber Insulin für individueller Ausprägung führt. Bei ca. 25–40 % der Mädchen
die metabolischen Spätfolgen dieser intrauterin beginnenden mit Ullrich-Turner-Syndrom liegen zudem angeborene Ano-
Wachstumsstörung verantwortlich ist [44]. Diese Insulinresis- malien im Bereich der Nieren vor, wobei hier Hufeisennieren,
tenz scheint sich schon in den ersten zwei Lebensjahren, also Rotationsanomalien und einseitige Nierenaplasien im Vorder-
in der Phase des sog. „catch-up growth“, zu entwickeln. Durch grund stehen. Die chromosomale Störung kann komplett oder
Adipositas wird dieser Effekt noch verstärkt [45]. SGA-ge- als Mosaik vorliegen. Da der Genotyp wesentlich den Phä-
borene Kinder ohne „catch-up growth“ zeigen im Gegensatz notyp prägt, können die als typisch beschriebenen klinischen
dazu eine normale Insulinsensitivität im Vergleich zu AGA- Stigmata nur sehr diskret vorliegen oder vollständig fehlen.
Kindern („appropriate for gestational age“). Unter der The- Richtungsweisend sind in der Anamnese eines kleinwüchsi-
rapie mit rhGH kommt es bei diesen Kindern zu einer vor- gen Mädchens postpartal beobachtete Hand- und/oder Fußrü-
übergehenden, reversiblen Erhöhung der Insulinspiegel ohne ckenödeme und rezidivierende Otitiden aufgrund der Enge der
nachweisliche negative Langzeiteffekte auf den Kohlenhy- Gehörgänge. Bei jedem Mädchen mit unklarem Kleinwuchs
dratstoffwechsel. Insgesamt sprechen die bis heute vorliegen- ist daher eine Chromosomenanalyse zu empfehlen. Gelegent-
den Ergebnisse verschiedener Untersuchungen durchwegs lich kann bei betroffenen Mädchen Y-chromosomales Mate-
für eine positive Beeinflussung der Stoffwechselprozesse bei rial nachgewiesen werden, wobei in diesen Fällen ein erhöh-
SGA-Kindern durch die Therapie mit rhGH. Besonders unter tes Gonadoblastomrisiko in den dysgenetischen Gonaden vor-
dem Gesichtspunkt eines erhöhten Risikos von SGA-Kindern liegt und daher präpubertär eine prophylaktische Gonadekto-
zur Entwicklung eines metabolischen Syndroms im Erwach- mie empfohlen wird [46].
senenalter und dem dadurch bedingten, durch wissenschaftli-
che Studien belegten erhöhten Risikos für das Auftreten von Der Kleinwuchs bei Mädchen mit UTS ist typischerweise dis-
nichttödlichen Herz-Kreislauf-Erkrankungen darf die rhGH- kret disproportioniert mit verkürzten Extremitäten im Ver-
Therapie nicht nur unter dem Aspekt der Verbesserung der Er- gleich zur Sitzhöhe. Verantwortlich hierfür ist der heterozy-
wachsenengröße betrachtet werden. Die Hypothese einer prä- gote Verlust des SHOX-Gens („short stature homeobox-con-
J KLIN ENDOKRINOL STOFFW 2015; 8 (1) 11Kleinwuchs – Differenzialdiagnose und therapeutische Optionen
Erste Berichte über eine positive Beeinflussung des Wachs-
tums bei Mädchen mit UTS stammen aus den 1980er-Jahren
[48]. Der Beginn einer wachstumsfördernden Therapie mit
rhGH in einer supraphysiologischen/pharmakologischen Do-
sierung von 45–50 µg/kg/Tag wird in der Regel ab einem Al-
ter von 4–5 Jahren empfohlen. Berichte, dass ein deutlich frü-
herer Therapiebeginn (9 Monate bis 4 Jahre) mit einem deut-
lich größeren Erfolg der Therapie verbunden ist, sind durch
Endlängendaten noch nicht belegt [49]. Ziel der Therapie mit
rhGH ist eine Verbesserung der Endgröße der Mädchen um
ca. 5–7 cm, wobei das Ansprechen auf die Therapie im 1. Be-
handlungsjahr den größten positiv prädiktiven Wert zur Ab-
schätzung des Therapieerfolges darstellt [31, 50]. Der wachs-
tumsfördernde Effekt der Therapie ist dosisabhängig, dennoch
sind tägliche Dosen > 50 µg/kg aufgrund des nicht abschätz-
baren Langzeitrisikos nicht zu empfehlen. Neben der wachs-
Abbildung 4: Madelung-Deformität klinisch und radiologisch. Die Veränderung ist
charakteristisch für kleinwüchsige Kinder mit Defekten im Bereich des SHOX-Gens tumsfördernden Therapie ist die zeitgerechte Induktion der
(„short stature homeobox-containing gene“), z. B. Leri-Weill-Dyschondrosteosis. Ne- Pubertätsentwicklung durch die Gabe von Östrogenen für die
ben einer vermehrten Krümmung des Radius kommt es zu einer distalen Subluxation betroffenen Mädchen von besonderer Bedeutung. Regelmäßi-
der Ulna nach dorsal (Bajonett-Zeichen) mit konsekutiver Supinationshemmung im
Handgelenk. Eine Madelung-Deformität kann auch bei Mädchen mit Ullrich-Turner- ge und engmaschige kardiologische Kontrollen sind fixer Be-
Syndrom auftreten. standteil in der Betreuung von Mädchen mit UTS [51, 52].
taining gene“) [47]. Die Geburtslänge ist meist normal, die Genetische, primäre chondroossäre Wachs-
Wachstumsstörung wird bereits in den ersten Lebensjahren tumsstörungen
mit einem kontinuierlichen Abfall von der Perzentile nach un- Primäre chondroossäre Kleinwuchsformen werden in Skelett-
ten manifest. Aufgrund der ovariellen Insuffizienz ist der pu- dysplasien (Defekte des Knochens als Gewebe) und Dysosto-
bertäre Wachstumsschub gegenüber gesunden Mädchen um sen (Defekte des Knochens als Organ) unterteilt [53]. Aktu-
ca. 5–10 cm geringer. Die Endgröße der Mädchen liegt unbe- ell umfasst die Klassifikation 40 verschiedene Gruppen. Der
handelt ca. 20 cm unter der genetischen Zielgröße. Kleinwuchs ist bei 80–90 % der Skelettdysplasien das Leit-
Ausgeprägter Kleinwuchs < –3 SD (1 Messung) oder < –2,5 SD ( 2 Messungen)
Abweichung zur genetischen Ziellänge > –2 SD
Wachstumsabfall > –1 SD = Wachstumsgeschwindigkeit < P25 bei Kindern ab dem 4. Lebensjahr
Proportionierter KW Disproportionierter KW
SGA mit fehlendem
– Ullrich-Turner-
Aufholwachstum
Syndrom
PWS IGF-1 Ļ IGF-1 nl/Ļ
CRI IGF-BP3 Ļ IGF-BP3 nl GH-Therapie
– Skelettröntgen
GH-Therapie – ev. Molekulargenetik
(SHOX, FGFR3, PSACH)
– Stoffwechseldiagnostik
Retardiertes Knochenalter Altersentspr. Knochenalter
Konstitutionelle Verzögerung Familiärer Kleinwuchs
Wachstumshormontestung
von Wachstum und Pubertät Idiopathischer Kleinwuchs
Leri-Weill-Dyschondrosteosis
A-/Hypochondroplasie
1. GH-Test Ĺ und
Pseudoachondroplasie
IGF-1/ IGF-BP3 Ļ
Speicherkrankheiten
< –2,5 SDS 1. GH-Test Ļ 1. GH-Test nl Verlaufskontrolle 6–12 Monate
IGF-1-Generationstest GH-Therapie
mit fehlendem Anstieg Pathologisches nächtliches GH- oder andere
2. GH-Test Ļ 2. GH-Test nl
Sekretionsprofil Therapie
SPIGFD
GH-Mangel
Neurosekretorische Dysfunktion
MRI der Hypophyse
Kombinierte Ausfälle ?
IGF-1-Therapie (GH-1, Pit-1, PROP-1, HESX-1) GH-Therapie
Abbildung 5: Stufenschema zur Abklärung von Wachstumsstörungen. Aus der Leitlinie „Abklärung Kleinwuchs/Diagnostik des Wachstumshormonmangels im Kindes- und
Jugendalter“ der Arbeitsgruppe Pädiatrische Endokrinologie und Diabetologie (APEDÖ) der Österreichischen Gesellschaft für Kinder- und Jugendheilkunde (ÖGKL). Stand 2009.
Mit freundlicher Genehmigung der APEDÖ.
BP3: „binding protein“ 3; CRI: „chronic renal insufficiency“; GH: „growth hormone“; IGF: „insulin-like growth factor“; PWS: Prader-Willi-Syndrom; SGA: „small for gestational age“
12 J KLIN ENDOKRINOL STOFFW 2015; 8 (1)Kleinwuchs – Differenzialdiagnose und therapeutische Optionen
symptom und kann bereits intrauterin oder erst postnatal auf- der klinisch häufig schwer zu diagnostizierenden HCH wichti-
treten. Skelettdysplasien können primär vorliegen oder Teil ge Hinweise liefern. Kinder mit ACH und HCH weisen meist
eines komplexen genetischen Syndroms sein (u. a. Ullrich- Geburtsmaße im unteren Normalbereich auf. Besonders in
Turner-Syndrom, Noonan-Syndrom, Silver-Russel-Syndrom, den ersten drei Lebensjahren bleiben die Kinder deutlich im
3M-Syndrom, DiGeorge-Syndrom etc.). Klinisch charakteri- Wachstum zurück, die Erwachsenengröße liegt bei Kindern
siert sind Skelettdysplasien durch eine mehr oder weniger aus- mit ACH im Bereich zwischen 116 und 133 cm bei Frauen,
geprägte Disproportion. Neben dem Rumpfkleinwuchs (spon- zwischen 120 und 142 cm bei Männern und bei Kindern mit
dylo-epiphysäre Dysplasie) und der Asymmetrie (Silver-Rus- HCH zwischen 128 und 165 cm. Zahlreiche Therapieversuche
sel-Syndrom) werden akromele (Hand/Fuß verkürzt; z. B. tri- mit rhGH zeigten ein sehr individuelles Ansprechen auf die
chorhinophalangeales Syndrom) von mesomelen (Unterarm/ Therapie, wobei positive Effekte vor allem in den ersten zwei
Unterschenkel verkürzt; z. B. Leri-Weill-Dyschondrosteosis, Behandlungsjahren beobachtet wurden [57]. Die Körper-
Ellis-van-Crefeld-Syndrom) und rhizomelen (Oberarm/Ober- proportionen wurden unter der Therapie nicht negativ beein-
schenkel verkürzt; z. B. Typ Patterson-Lowry) Phänotypen flusst. Dennoch kann aufgrund fehlender Daten zur Endgröße
unterschieden. Die genetische Evaluation der verschiedenen der Patienten unter Therapie keine generelle Empfehlung ab-
Kleinwuchsformen wurde rezent von Zabel und Lausch in der gegeben werden. Erfolgversprechender scheint ein Therapie-
Monatsschrift Kinderheilkunde dargestellt [54]. Im Kontext ansatz mit einem langwirksamen Agonisten (BMN-111) des
einer ausführlichen Familienanamnese und einer klinischen Typ-C-natriuretischen Peptids (CNP) durch Blockierung des
Fotodokumentation liefern Röntgenaufnahmen von Teilen des bei ACH überaktivierten MAP-Kinase-Signalweges [58]. Im
Skeletts oder ein gesamter Skelettstatus, und hier besonders Tierexperiment konnten mit dieser Therapie eine Aufhebung
die Beurteilung der Epiphysenfugen, meist die entscheiden- der Zellausreifungsstörung in der Epiphyse und eine Verbes-
den Hinweise für die Zuordnung zu einer bestimmten Krank- serung des Wachstums und des rhizomelen Phänotyps beob-
heitsentität. achtet werden. Die Daten konnten in Deutschland in einer
noch nicht abgeschlossenen Phase-II-Studie an Kindern be-
Nur ein geringer Teil der primären Wachstumsstörungen stätigt werden.
spricht auf pharmakologische Dosen einer wachstumsför-
dernden Therapie an. So werden beispielsweise Patienten mit
Silver-Russel-Syndrom seit Jahren unter der Indikation ei- Relevanz für die Praxis
ner SGA-Konstellation bei Geburt mit rhGH behandelt [55]. Ein Kleinwuchs per se stellt noch keine Krankheitsenti-
Eine eigenständige Indikation zur Therapie mit rhGH stel- tät dar und muss von einer Wachstumsstörung mit perzen-
len seit 2007 Kinder mit SHOX-Mutationen dar, da bei die- tilenflüchtigem Wachstum nach unten abgegrenzt wer-
sen Patienten eine vergleichbare Wirksamkeit der Therapie den. Die Differenzialdiagnose umfasst eine Vielzahl von
wie bei Mädchen mit UTS nachgewiesen wurde [56]. Die Be- Krankheitsbildern. Die Abklärung erfordert ein struktu-
handlung erfolgt wie beim UTS durch die tägliche s.c. rhGH- riertes Vorgehen nach von Fachgesellschaften erstellten
Injektion von pharmakologischen Dosen von 45–50 µg/kg. Stufenschemata und eine gute Kooperation zwischen be-
Zum klinischen Spektrum dieser Erkrankung gehören Kinder treuenden Ärzten und Spezialambulanzen für pädiatrische
mit einer autosomal-dominant vererbten Leri-Weill-Dyschon- Endokrinologie (Abb. 5). Der klinischen Befunderhebung
drosteosis, die klinisch neben dem mesomelen Kleinwuchs kommt die zentrale Rolle zu, die laborbiochemischen und
durch das Vorliegen einer vermehrten Krümmung des distalen molekulargenetischen Untersuchungen dienen der Bestä-
Radius und einer Madelung-Deformität charakterisiert sind tigung. Die Differenzierung zwischen einer proportio-
(Abb. 4). Diese Veränderung mit einer distalen Subluxation nierten und einer disproportionierten Wachstumsstörung
der Ulna nach dorsal (Bajonett-Zeichen) mit konsekutiver Su- liefert den entscheidenden Hinweis, ob von einer primären
pinationshemmung im Handgelenk kann auch bei Mädchen oder sekundären Wachstumsstörung auszugehen ist. Bei
mit UTS angetroffen werden. Weitere schwere Formen aus der weiteren Unterteilung von sekundären Wachstumsstö-
der SHOX-Kleinwuchsgruppe sind das Xp-Mikrodeletions- rungen lag der Fokus aufgrund der therapeutischen Optio-
syndrom und die mesomele Dysplasie Typ Langer. Neben he- nen > 40 Jahre lang am Wachstumshormon. Die Verfüg-
terozygoten und homozygoten Punktmutationen und Deletio- barkeit von rekombinantem IGF-1 zur Therapie schwe-
nen des SHOX-Gens werden in diese Gruppe auch Deletionen rer Kleinwuchsformen macht ein Umdenken erforder-
der pseudoautosomalen Region auf dem X- oder Y-Chromo- lich. Wenn auch ähnlich arbiträr wie die Unterscheidung
som (PAR1) und Deletionen verschiedener SHOX-regulatori- in „Kleinwuchs mit/ohne Wachstumshormonmangel“, er-
scher Sequenzen auf dem X-Chromosom subsummiert. scheint eine Differenzierung in „Kleinwuchs mit primä-
rem/sekundärem IGF-1-Mangel“ im Hinblick auf die the-
Der letale thantophore Kleinwuchs (TD) gehört zusammen rapeutischen Optionen sinnvoll.
mit der Achondroplasie (ACH) und der mildesten Form der
Hypochondroplasie (HCH) in die „Fibroblast growth factor
receptor 3“- (FGFR3-) Familie der ossären Kleinwuchsfor-
men. Klinisch sind diese Skelettdysplasien durch eine rhi- Interessenkonflikt
zomele Verkürzung der Extremitäten, einen relativ langen
Rumpf und Fehlbildungen des Handskeletts sowie des Rip- K. K. ist Mitglied der Expertengruppe „Short Stature“ der Fir-
penthorax charakterisiert. Ein Röntgen der Lendenwirbelsäu- ma Ipsen, einem Hersteller von IGF-1, und Ko-Autor der „En-
le a.p. und seitlich mit Nachweis einer fehlenden Progredienz dologic Library“ der Firma Merck Serono. K. K. erhielt zudem
der Interpedunkularabstände von kranial nach kaudal kann bei Vortragshonorare von den Firmen Pfizer und Novo Nordisk.
J KLIN ENDOKRINOL STOFFW 2015; 8 (1) 13Sie können auch lesen

















































