Neurodegenerative Erkrankungen des Kindesalters - De Gruyter
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Schwerpunktthema: Genetik der neurodegenerativen Erkrankungen
medgen 2018 · 30:231–237 Angela Schulz · Miriam Nickel
https://doi.org/10.1007/s11825-018-0194-2 Klinik für Kinder- und Jugendmedizin, Universitätsklinikum Hamburg-Eppendorf, Hamburg, Deutschland
Online publiziert: 18. Juli 2018
© Der/die Autor(en) 2018
Neurodegenerative
Erkrankungen des Kindesalters
Einführung gen Fällen aber auch des Sehvermögens Bei den NCL wird lysosomales Spei-
(hier sind insbesondere die NCL-Krank- chermaterial –sogenanntes Ceroid-Lipo-
Zu den neurodegenerativen Erkrankun- heiten zu nennen) und selten auch des fuszin – nicht mehr abgebaut. Die Spei-
gen des Kindesalters, die vor allem das Hörvermögens (einige Formen der MPS cherung solcher Substanzen führt in Ner-
zentrale Nervensystem (ZNS) betreffen, und Leukodystrophien). venzellen des Gehirns zu deren Abster-
gehören vorwiegend die neuronalen Ce- Aufgrund abweichender genetischer ben.
roid-Lipofuszinosen (NCL), einige For- Ursachen gibt es auch innerhalb der
men der Mukopolysaccharidosen (MPS) Krankheitsgruppen viele Unterschie- Neue Nomenklatur
sowie der Leukodystrophien (. Tab. 1). de hinsichtlich Krankheitsbeginn und von NCL-Erkrankungen
Hierbei handelt es sich bei den NCL- -verlauf – beispielsweise bezüglich Aus-
Krankheiten als Gruppe um die am häu- prägung und Reihenfolge der Symptome Traditionell wurden die NCL-Krankhei-
figsten auftretenden Formen. Außerdem [2–4]. ten nach dem Erkrankungsalter in ange-
gibt es weitere Stoffwechselkrankheiten Auch wenn die überwiegende Mehr- borene, infantile, spätinfantile, juvenile
aus der Gruppe der Eisenspeichererkran- zahl der neurodegenerativen Erkran- und adulte Formen eingeteilt. In jünge-
kungen, der Aminoazidopathien, der Or- kungen im Kindesalter monogenen rer Zeit wurde deutlich, dass die NCL-
ganoazidopathien und der Harnstoffsyn- Ursprungs ist, gibt es multifaktorielle Krankheiten jedoch deutlich heterogener
thesestörungen, die ebenfalls zu Neuro- und erworbene Krankheiten: Zu den sind als bisher angenommen und zudem
degeneration bei Kindern führen [1]. seltenen multifaktoriellen neurodegene- Mutationen im selben Gen zu sehr unter-
Alle diese Stoffwechselkrankheiten rativen Erkrankungen im Kindesalter schiedlichen Phänotypen und klinischen
haben gemeinsam, dass die Betroffenen zählen unter anderem eine früh be- Verläufen führen können. Aus diesem
initial vollkommen gesund erscheinen – ginnende multiple Sklerose oder eine Grund wurde die bisherige Nomenkla-
mit Ausnahme der kongenitalen CLN10- amyotrophe Lateralsklerose. Des Weite- tur durch eine international entwickel-
Form. Jedoch kommt es dann im Kindes ren gibt es seltene erworbene Formen te kombinierte genetische und klinische
– oder Jugendalter zu einem unauf- wie z. B. eine subakute sklerosierende Klassifikation der NCL-Krankheiten er-
haltsam fortschreitenden Verlust bereits Panenzephalitis, HIV oder ein nutritiver setzt (. Tab. 1). Diese Nomenklatur klas-
erlernter kognitiver Fähigkeiten, wes- Vitamin B12-Mangel. sifiziert sowohl das defekte Gen als auch
halb diese Krankheiten auch oft unter Im Folgenden soll eine Übersicht über das Alter bei Krankheitsbeginn (konge-
dem Begriff „Kinderdemenz“ zusam- die häufigsten monogenen neurodegene- nital, infantil, spätinfantil, juvenil oder
mengefasst werden. Die Mehrzahl dieser rativen Erkrankungen des ZNS im Kin- adult) [5].
Krankheiten gilt als progredient und desaltergegebenwerden, inderÜbersicht
unheilbar und führt zu einem frühzeiti- in . Abb. 1 dargestellt. Besonderer Fokus Das genetische Spektrum
gen Tod des Kindes, zum Teil innerhalb wird hier auf die häufigste Gruppe, die der NCL-Krankheiten
weniger Jahre nach dem Auftreten ers- NCL, gelegt.
ter Symptome. Sie sind alle genetisch Die NCL-Krankheiten sind monogene
bedingt mit in der Mehrzahl autosomal- Neuronale Ceroid- Krankheiten, sodass jede Form tatsäch-
rezessiver Vererbung. Lipofuszinosen (NCL) lich eine separate Krankheitsentität dar-
Ursächlich bei all diesen Krankheiten stellt. Eine wachsende Anzahl von Genen
sind genetische Defekte, die durch Spei- Bei den NCL handelt es sich um ly- wird als Ursache identifiziert. Alle kind-
cherung von nicht abbaubaren Substra- sosomale Speicherkrankheiten und die lichen Formen sind autosomal-rezessiv
ten zu einem Absterben von Nervenzel- häufigste Ursache für Kinderdemenz vererbt mit Ausnahme einer extrem sel-
len und damit irreversiblen Hirnschäden [1]. Betroffen sind dabei vorwiegend die tenen Art von adulter NCL. Bis heute
führen. graue Substanz des cerebralen Cortex wurden 14 verschiedene NCL-Formen
Die Folge ist ein progredienter Verlust (. Abb. 2) und die Retina (. Abb. 3; [4]). beschrieben (. Tab. 1; [6–11]). Es müssen
psychomotorischer Fähigkeiten, in eini- jedoch noch mehr NCL-Gene existieren,
medizinische genetik 2 · 2018 231Schwerpunktthema: Genetik der neurodegenerativen Erkrankungen
Tab. 1 Korrelation von Genotyp und Phänotyp in NCL
Gen Protein Anzahl Genotyp-Phänotyp- Krankheits- Klassische erste Ultrastructural
Mutationen Korrelation beginn Symptome Pathology
CLN1/PPT1 Lysosomales 71 Infantil 6–12 Mo Stagnation/Rückschritte Granular osmiophilic
Enzym Spätinfantil 1,5–4 J der psychomotorischen deposits (GROD)
Entwicklung
Juvenil 5–7 J
Adult >16 J
CLN2/TPP1 Lysosomales 121 Spätinfantil 2–4 J Epilepsie, verzögerte Curvilinear profiles (CL)
Enzym Juvenil 8J Sprachentwicklung,
Ataxie
Verzögert –
SCAR7 >11 J
CLN3 Lysosomales 78 Juvenil 4–7 J Visusverlust Fingerprint profiles (FP)
Membranprotein Verzögert
Autophagische vakuoläre
Myopathie
Retinitis pigmentosa
Stäbchen-Zapfen-Dystrophie
CLN4/ Cytoplasmatisches 2 Adult >20 J Epilepsie, Ataxie, Ver- Diverse and often
DNAJC5 lösliches Protein autosomal dominant haltensauffälligkeiten mixed
(Parry-Krankheit)
CLN5 Lysosomales 37 Spätinfantil 4–5 J Verzögerung der Rectilinear profiles (RL)
Enzym Juvenil 5–7 J psychomotorischen & Condensed storage
Entwicklung, inclusions
Verzögert – Visusverlust
Adult >16 J
CLN6 Endoplasmatisches 70 Spätinfantil 18 Mo+ Epilepsie und Rectilinear profiles (RL)
Retikulum Verzögert – motorischer Abbau & Condensed storage
Membranprotein inclusions
„Adult Kufs Typ A“ >16 J
Juvenile cerebelläre Ataxie –
CLN7/MFSD8 Lysosomales 38 Spätinfantil 1,5–6 J Epilepsie, Rectilinear profiles (RL)
Membranprotein Juvenil verzögert >7 J psychomotorischer & Condensed storage
Abbau inclusions
Makuladystrophie –
Stäbchen-Zapfen-Dystrophie –
CLN8 Endoplasmatisches 35 Spätinfantil 2–7 J Epilepsie Rectilinear profiles (RL)
Retikulum Verzögert 5–10 J & Condensed storage
Membranprotein inclusions
„EPMR/Northern epilepsy“ –
CLN10/CTSD Lysosomales 12 Kongenital Pre-/ Epilepsie, Spastik, Granular osmiophilic
Enzym perinatal zentrale Atem- deposits (GROD)
Spätinfantil 4J regulationsstörung
Juvenil 8–15 J
Adult >20 J
CLN11/GRN Sezerniertes 2 Adult >20 J Schnell fortschreitender Rectilinear profiles (RL)
Protein Frontotemporale Demenz Visusverlust, Epilepsie
(bei Heterozygotie)
CLN13/CTSF Lysosomales 11 „Adult Kufs Typ B“ >20 J Tremor, Ataxie, Fingerprint profiles (FP)
Enzym Epilepsie
Varianten in weiteren Genen wurden vereinzelt mit NCL-ähnlichen Phänotypen verbunden:
CLN12/ATP13A2: Mutationen verursachen normalerweise das Kufor-Rakeb Syndrom, wurden aber auch bei Patienten mit juvenilem NCL-Phänotyp
beschrieben,
CLN14/KCTD7: Mutationen verursachen eine progressive Myoklonus-Epilepsie oder ein „Opsoclonus-myoclonus-ataxia-like syndrome“, aber werden
auch mit infantiler und spätinfantiler NCL in Verbindung gebracht
Fett gedruckt: „klassischer“ häufigster Phänotyp; kursiv gedruckt: Nicht-NCL-Phänotypen, die in manchen Fällen typischer für Mutationen in diesem Gen
sind
PPT1 „Palmitoyl protein thioesterase 1“, TPP1 „Tri-peptidyl peptidase 1“, DNAJC5 „DNAJ homolog subfamily C member 5“ (auch „cysteine string
protein alpha“ oder CSPa), MFSD8 „Major facilitator superfamily domain-containing protein 8“, CTSD Cathepsin D, GRN Granulin; CTSF Cathepsin F,
SCAR7 „Spinocerebellar Ataxia, Autosomal Recessive 7“, EPMR „Epilepsy, Progressive, With Mental Retardation“
232 medizinische genetik 2 · 2018Zusammenfassung · Abstract
da bei einigen Patienten Mutationen in medgen 2018 · 30:231–237 https://doi.org/10.1007/s11825-018-0194-2
keinem der bekannten NCL-Gene nach- © Der/die Autor(en) 2018
gewiesen werden können, obwohl sie ty-
pische NCL-Symptome und charakteris- A. Schulz · M. Nickel
tisches lysosomales Speichermaterial in Neurodegenerative Erkrankungen des Kindesalters
ihren Zellen aufweisen. Viele der Gene
kodieren für scheinbar in ihrer Funkti- Zusammenfassung
Das Verständnis der neurodegenerativen zu einer Degeneration des zentralen
on vollkommen unterschiedliche Prote-
Erkrankungen im Kindesalter hat sich in Nervensystems führen, die neuronalen
ine wie lysosomale Enzyme (z. B. CLN1, jüngster Zeit rasant verändert: Nicht nur Ceroid-Lipofuszinosen (NCL). Die Anzahl der
CLN2, CLN5, CLN10 und CLN13) und die Anzahl unterschiedlicher Krankheiten NCL-verursachenden Gene und das Wissen
Membranproteine, die in verschiedenen und zugrunde liegender Gendefekte nimmt über Genotyp-Phänotyp-Korrelationen
Organellen lokalisiert sind, einschließ- stetig zu, auch die Ansätze für Diagnostik sind in den letzten Jahren gewachsen und
und Therapie haben sich aufgrund neuerer erste Therapien wurden entwickelt. Damit
lich dem Lysosom (z. B. CLN3, CLN6,
technologischer und therapeutischer stellt diese Krankheitsgruppe die schnelle
CLN7 und CLN8). Auch Mutationen ei- Fortschritte in dieser Krankheitsgruppe wissenschaftliche Entwicklung auf dem
ner APTase (CLN12) [10] oder eines Ka- weiterentwickelt. Es wurden neue Gendefekte Gebiet der seltenen, neurodegenerativen
liumkanals (CLN14) [12] scheinen eine identifiziert, die eine Grundlage für das Erkrankungen im Kindesalter sehr gut dar.
NCL-Erkrankung zu verursachen. Das Verständnis der molekularen Mechanismen,
die dieser Krankheitsgruppe zugrunde Schlüsselwörter
CLN4-Gen (auch DNAJC5 genannt) ko-
liegen, sowie für die Entwicklung gezielter Neurodegeneration · Neuronale Ceroid-
diert für ein Protein mit mutmaßlicher Therapien bieten. Diese Übersichtsarbeit Lipofuszinosen · Leukodystrophien ·
Funktion in Synapsen [11]. Für keines der konzentriert sich hauptsächlich auf eine Mucopolysaccharidosen · Kinderdemenz
bekannten NCL-Proteine konnte bisher der häufigsten Krankheitsgruppen, die
die genaue Fehlfunktion aufgeklärt wer-
den, die zur Neurodegeneration führt.
Neurodegenerative diseases of childhood
Die klinischen Phänotypen der Abstract
NCL-Krankheiten The understanding of neurodegenerative neuronal ceroid lipofuscinosis (NCL). The
diseases of childhood has been changing number of NCL-causing genes and knowledge
Trotz ihrer genetischen Heterogenität ha- rapidly in recent times: not only is the number about genotype–phenotype correlations has
ben die NCL-Erkrankungen klinisch in of different diseases and underlying genetic been growing over the past few years and the
den meisten Fällen eine Kombination defects steadily increasing, approaches first therapies have been developed. Hence,
to diagnosis and treatment have also this group of diseases represents the rapid
aus psychomotorischem Abbau, Epilep- scientific development in the field of rare
developed because of recent technological
sie und Visusverlust gemeinsam. and therapeutic advances relating to this neurodegenerative diseases in childhood very
Den meisten NCL-Genen wird ein group of disorders. New gene defects have well.
klar erkennbarer „klassischer“ Krank- been identified that provide a basis for
heitsphänotyp zugeordnet, der mit einem understanding the molecular mechanisms Keywords
underlying this group of diseases, and for Neurodegeneration · Neuronal ceroid
vollständigen Funktionsverlust des mu- lipofuscinoses · Leukodystrophies ·
the development of targeted therapies. This
tierten Proteins vereinbar ist, am ehesten review focuses predominantly on one of the Mucopolysaccharidoses · Childhood
verursacht durch intrazelluläre Fehllo- most common groups of diseases leading to dementia
kalisation oder frühzeitigen Abbau. Je- degeneration of the central nervous system,
doch werden vermehrt Patienten mit
„atypischen“ Phänotypen identifiziert,
verursacht durch sogenannte „milde“
Mutationen, die nicht zu einem vollstän- alle Formen gemeinsam, mit Ausnahme ab, die das Alter beim Ausbruch und
digen Funktionsverlust des zugehörigen einer seltenen angeborenen Form, dass den Krankheitsphänotyp beeinflussen.
Proteins führen (. Tab. 1). vor Beginn der ersten Symptome eine Erste Symptome bei einem infantilen
Die zunehmende Implementierung normale psychomotorische Entwick- Phänotyp (Erkrankungsalter 6–24 Mo-
von „Next Generation Sequencing Pa- lung vorliegt. Die Hauptalarmsymptome nate) und auch einem spätinfantilen
nels“ und Exom-Sequenzierung als es- sind die Kombination von mindestens Phänotyp (Erkrankungsalter 2–5 Jah-
senzielle diagnostische Werkzeuge zur zwei Symptomen wie Demenz, Epilep- re) verlangsamen die psychomotorische
Beurteilung seltener Erkrankungen führt sie, motorische Verschlechterung und Entwicklung. Dem folgt rasch ein Ent-
zu mehr Diagnosen von NCL-Patien- Sehverlust. Das Alter bei Krankheits- wicklungsstillstand, dann später Verlust
ten einschließlich jener, die von den beginn kann von der Geburt bis zum der psychomotorischen Fähigkeiten und
klassisch anerkannten Phänotypen ab- Erwachsenenalter reichen. Die Reihen- Beginn der Epilepsie. Diese Regressi-
weichen. Diese Patienten hätten zuvor folge, in der Symptome auftreten, ist on der psychomotorischen Fähigkeiten
möglicherweise niemals eine genetische variabel und hängt von der Kombinati- wird oft als eine Nebenwirkung von An-
NCL-Diagnose erhalten. Dennoch haben on der zugrunde liegenden Mutationen tiepileptika falsch interpretiert, was die
medizinische genetik 2 · 2018 233Schwerpunktthema: Genetik der neurodegenerativen Erkrankungen
Neuronale Ceroid- Mukopolysaccharidosen
Lipofuszinosen (NCL) (MPS)
N =14 N=6
Neurodegenerative
ZNS-Krankheiten im
Kindesalter
Leukodystrophien Andere
N >20 Stoffwechselkrankheiten
Abb. 1 8 Übersicht über Krankheitsgruppen, die Degeneration des zentra-
len Nervensystems im Kindesalter verursachen
Diagnose dieser sich schnell verschlech-
ternden NCLs verzögert. Sehverlust tritt
erst in den späten Stadien dieser Krank-
heiten auf. Im Gegensatz dazu sind
die ersten Symptome im jugendlichen Abb. 2 8 Cerebrale Magnetresonanz-Tomographie (T2-Wichtung) bei
Phänotyp (Erkrankungsalter 5–7 Jahre) einer Patientin mit spätinfantiler CLN2-Krankheit. a Alter 3,5 J. b Alter 5 J.
Eine progrediente Atrophie der cerebralen und cerebellären grauen Sub-
meist Sehverlust, gefolgt von Demenz stanz ist sichtbar
und Verhaltensänderungen, dann Ver-
lust der motorischen Fähigkeiten und
Epilepsie in den frühen Teenagerjahren. -schweregrad bei jungen, spätinfantilen erst in späten Krankheitsstadien evident,
In adulten Phänotypen fehlt der Seh- CLN2-Patienten bis in die späten Krank- während er bei juvenilen Patienten in der
verlust gewöhnlich. Patienten mit pro- heitsstadien. Bei jungen CLN1-Patienten Regel das Erstsymptom bei sonst voll-
gressiver Myoklonus-Epilepsie (Typ A) neigt die Anfallshäufigkeit jedoch in den kommen gesund erscheinendem Patien-
oder Demenz mit motorischem Verfall späteren Krankheitsstadien dazu abzu- ten darstellt [4].
(Typ B) zeigen typischerweise einen nehmen, und bei jugendlichen CLN3-Pa- Das klinische Spektrum der NCL-
Erkrankungsbeginn um das Alter von tienten sind Anfälle selten, wobei sich mit Krankheiten hat sich neben den „klassi-
30 Jahren, aber dies kann vom Teenager dem Alter nur eine leichte Verschlechte- schen“ oben beschriebenen Phänotypen
bis zu den Fünfzigern reichen. rung zeigt [4]. Die motorischen Sympto- auch um „atypische“ Phänotypen er-
Auch wenn die verschiedenen NCL- me umfassen Ataxie (einschließlich Dys- weitert: Bei manchen Patienten kann
Phänotypen ähnliche klinische Merkma- metrie, Dysarthrie), Dysphagie, Myoklo- eines der NCL-Hauptsymptome vor-
le aufweisen, unterscheidet sich ihr klini- nus, Chorea, Tremor und Dystonie, be- herrschend sein, während andere fehlen:
scher Schweregrad und Phänotyp sogar sonders bei infantilen und spätinfantilen Zum Beispiel gibt es bei der CLN2-
für den gleichen genetischen Typ. Eine Patienten. Andere umfassenParkinsonis- Erkrankung den SCAR7-spinozerebellä-
Verzögerung der expressiven Sprachent- mus, insbesondere bei juveniler CLN3- ren Ataxie-Phänotyp, bei dem Patienten
wicklung hat sich bei 83 % der CLN2-Pa- Erkrankung, und einige stereotype Be- hauptsächlich an Ataxie leiden und
tienten als Vorläufer der Regression der wegungen, die bei verschiedenen Arten keine Epilepsie oder Sehverlust aufwei-
psychomotorischen Funktion erwiesen von NCL mit spätinfantilem und juveni- sen sowie einen Erkrankungsbeginn im
und kann zur Verbesserung der Frühdia- lem Erkrankungsalter beobachtet wur- Jugendalter mit Überleben bis in die
gnose verwendet werden: Kinder mit ei- den. Die Behandlung der Bewegungs- vierte Dekade zeigen [14]. Außerdem
ner Kombination aus verzögerter Sprach- störungen ist eine große Herausforde- verursachen bestimmte Mutationen im
entwicklung und neu auftretenden epi- rung und erfordert sowohl Medikamen- CLN3-Gen einen milderen Phänotyp
leptischen Anfällen, für die es keine kla- te als auch ein multidisziplinäres Team einschließlich isolierter, nicht syndro-
re Ursache gibt, sollten auf CLN2 getes- aus Physiotherapeuten, Ergotherapeuten maler Retinadegeneration [15]. Einige
tet werden [13]. Epilepsie ist bei fast al- und Ärzten. Der Visusverlust, verursacht dieser Patienten mit CLN3-Mutationen
len NCL-Patienten therapieresistent, mit durch eine Retinopathie, wird bei infan- leiden unter Visusmangel, Krampfan-
besonders hoher Anfallshäufigkeit und tilen und spätinfantilen Patienten meist fällen, starker Beteiligung des Herzens,
234 medizinische genetik 2 · 2018Die Identifizierung von vakuolisierten
Lymphozyten, ein Merkmal der CLN3-
Erkrankung und einiger anderer lyso-
somaler Störungen, kann durch einen
Blutausstrich bestätigt werden (. Abb. 4).
Während das Speichermaterial extraze-
rebral bei kindlichen Formen der NCLs
leicht nachzuweisen ist, ist dies nicht un-
bedingt der Fall, wenn NCL im Erwach-
senenalter auftritt.
Therapie der NCL-Krankheiten
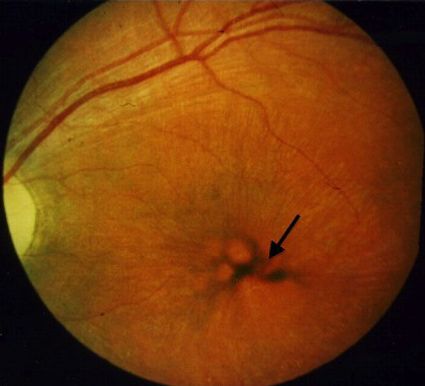
Abb. 3 8 Retinale Funduskopie eines 9-jähri- Abb. 4 8 Blutausstrich eines 9-jährigen juve-
gen juvenilen CLN3-Patienten: Irreguläre Pig- nilen CLN3-Patienten: Nachweis charakteristi- Bis vor Kurzem galten alle NCL-Krank-
mentablagerungen und verdünnte Gefäße als scher Speichervakuolen in Lymphozyten heiten als unheilbar und lediglich pallia-
klassische Hinweise für die Retinopathie tive Therapien standen zur Verfügung.
Die erste intracerebroventrikuläre En-
zeigen aber auch nach Jahrzehnten keine Diagnose der NCL-Krankheiten zymersatztherapie (ICV-ERT) für CLN2
motorische Verschlechterung. ist daher ein wichtiger Fortschritt und
Zusammenfassend wächst das Spek- Die Änderung diagnostischer Ansätze stellt die erste zugelassene Behandlung
trum verschiedener Phänotypen für unter Verwendung von NGS-Panels, die für eine der NCL-Formen dar [18]. Bei
NCL-Erkrankungen(. Tab. 1). BeiNCLs, entworfen wurden, um alle bekannten dieser Form der Enzymersatztherapie
die durch lysosomale Enzymdefekte wie NCL-Gene unter einer viel größeren werden in zweiwöchentlichen Abstän-
CLN1, CLN2 und CLN10 verursacht Gruppe von Störungen abzufragen (z. B. den 300 mg des rekombinanten Enzyms
werden, reicht das Spektrum vom infan- ein „Epilepsie“-Panel, „Retinopathie“- Tripeptidylpeptidase 1 (TPP1) über eine
tilen bis zum adulten Phänotyp. Zuneh- Panel), und auch unter Durchführung Rickham- oder Ommaya-Kapsel in die
mendes Wissen über den natürlichen von Exom-Sequenzierungen stellt nun Seitenventrikel des Gehirns appliziert.
Krankheitsverlauf der verschiedenen eine umfassende Testung in einem ein- Um die therapeutische Wirksamkeit
NCL-Formen (z. B. durch Sammeln sol- zigen Schritt sicher. Dies ist besonders zu testen, wurden Daten zum natürli-
cher Daten in internationaler Kooperati- nützlich für die genetische Diagnose chen Krankheitsverlauf, die im Rahmen
on [7]) hat gezeigt, dass für einige NCLs, atypischer Erkrankungen in einzelnen eines FP7-finanzierten europäischen
wie die spätinfantile CLN2-Erkrankung, Familien. Forschungskonsortiums DEM-CHILD
eine hohe Korrelation zwischen spezi- Neben der genetischen Diagnostik gesammelt wurden, als Vergleichsdaten
fischer Genmutation und klinischem stellt die Enzymtestung bei Verdacht verwendet [13]. Sicherheit und Wirk-
Phänotyp besteht, für andere der Phäno- auf CLN1- oder CLN2-Krankheit eine samkeit wurden in einer offenen Dosis-
typ-Schweregrad auch bei Patienten mit schnelle und kostengünstige Alternative Eskalationsstudie an 24 Patienten mit
identischem Mutationshintergrund, wie dar und sollte daher bei allen Kindern CLN2-Krankheit im Alter zwischen 3
bei der juvenilen CLN3-Erkrankung, mit frühkindlichem Erkrankungsbe- und 8 Jahren sowie einer Open-Label-
signifikant variieren kann. Darüber hi- ginn zunächst erwogen werden. Die Verlängerungsstudie getestet. Die pri-
naus können die Auswirkungen dieser Enzymaktivitätsmessung ist in einer mären Ziele waren die Bewertung der
Erkrankungen über das zentrale Ner- Trockenblutprobe möglich [17]. Fehlen- Sicherheit und Verträglichkeit der Thera-
vensystem und die Retina hinausgehen. de Aktivität des Enzyms PPT1 bestätigt pie sowie die Evaluation der Wirksamkeit
Eine kardiale Beteiligung bei adoles- die Diagnose CLN1-Krankheit, fehlen- anhand einer CLN2-krankheitsspezifi-
zenten und erwachsenen Patienten mit de Aktivität von TPP1 diejenige einer schen Ratingskala. In der Studie kam
CLN3-Erkrankung [16] deutet darauf CLN2-Krankheit. Eine entsprechende es bei 80 % aller behandelten Patienten
hin, dass es klinische Manifestationen Mutationsanalyse des jeweiligen Gens zu einer signifikanten Verzögerung des
außerhalb des ZNS gibt und dass andere sollte angeschlossen werden. Fortschreitens oder Stabilisierung der
Organsysteme ebenfalls betroffen sein Die elektronenmikroskopische ul- Krankheit, gemessen anhand der Bewer-
könnten, was eventuell erst bei erfolgrei- trastrukturelle Untersuchung auf lyso- tung der motorischen und sprachlichen
cher Therapie der ZNS-Beteiligung und somales Speichermaterial ist weiterhin Funktion nach 96 Behandlungswochen.
damit Verlängerung der Lebensdauer nützlich für die Bestätigung der NCL- Weitere, sich in Entwicklung und teil-
klinisch evident wird. Erkrankung, insbesondere für atypische weise bereits in klinischen Studien be-
Formen, die keine genetische Diagnose findliche Therapieoptionen für weitere
erhalten haben (. Tab. 1). In der Regel Formen der NCL-Krankheiten sind An-
wird dies mit einer Hautbiopsie oder sätze wie Gentherapie, Immunmodulati-
Blutprobe durchgeführt. on und Stammzelltherapie. . Tab. 2 gibt
medizinische genetik 2 · 2018 235Schwerpunktthema: Genetik der neurodegenerativen Erkrankungen
Tab. 2 Zusammenfassung abgeschlossener und laufender klinischer Therapiestudien bei NCL
Diagnose Titel Art der Therapie Phase Patienten- Studien- Status Clinical Trials
zahl dauer Number
CLN1 Cystagon to treat infantile neuronal Cystagon 4 9 2001–2013 Abgeschlossen NCT00028262
ceroid lipofuscinosis (a combination (Medikament)
therapy with Cystagon and N-Acetyl-
cysteine for INCL Patients)
CLN1 Study of human central nervous sys- Stammzellen 1 6 2006–2009 Abgeschlossen NCT00337636
und tem stem cells (HuCNS-SC) cells in
CLN2 patients with infantile or late infan-
tile neuronal ceroid lipofuscinosis
(NCL)
CLN2 Safety study of a gene transfer vec- AAV2CUhCLN2 1 10 2005–2019 Laufend, keine NCT00151216
tor for children with late infantile (intracerebrale Rekrutierung
neuronal ceroid lipofuscinosis Gentherapie)
CLN2 Safety study of a gene transfer vec- AAVrh.10CUhCLN2 1 25 2010–2016 Laufend, keine NCT01161576
tor (rh.10) for children with late (intracerebrale estimated to 2032 Rekrutierung
infantile neuronal ceroid lipofuscino- Gentherapie)
sis
CLN2 AAVrh.10 administered to children AAVrh.10CUhCLN2 1/2 8 2010–2022 Laufend, keine NCT01414985
with late infantile neuronal cero- (intracerebrale Rekrutierung
id lipofuscinosis with uncommon Gentherapie)
genotypes or moderate/severe im-
pairment
CLN2 Safety and efficacy study of BMN190 rhTPP1 BMN190 1/2 24 2013–2016 Abgeschlossen NCT01907087
for the treatment of CLN2 Patients. (intraventrikulä-
A phase 1/2 open-label dose-es- re Enzymersatz-
calation study to evaluate safe- therapie)
ty, tolerability, pharmacokine-
tics, and efficacy of intracerebro-
ventricular BMN 190 in patients
with late-infantile neuronal ceroid
lipofuscinosis (CLN2) disease
CLN2 A multicenter, multinational, exten- rhTPP1 BMN190 1/2 23 2015–2021 Laufend, keine NCT02485899
sion study to evaluate the long-term (intraventrikuläre Rekrutierung
efficacy and safety of BMN 190 in Enzymersatz-
patients with CLN2 disease therapie)
CLN2 A safety, tolerability, and efficacy rhTPP1 BMN190 2 15 2016–2023 Laufend, Ein- NCT02678689
study of intracerebroventricular BMN (intraventrikuläre schluss nur auf
190 in patients with CLN2 disease Enzymersatz- Einladung
therapie)
CLN3 Cellcept for treatment of juvenile Mycophenolate 2 19 2011–2015 Abgeschlossen NCT01399047
neuronal ceroid lipofuscinosis mofetil (Cellcept)
CLN6 Phase I/IIa gene transfer clinical trial scAAV9.CB.CLN6 1/2 12 2016–2019 Rekrutierung NCT02725580
for variant late infantile neuronal (Gentherapie) offen
ceroid lipofuscinosis, delivering the
CLN6 gene by self-complementary
AAV9
HuCNS-SC „human CNS stem cells“
eine aktuelle Übersicht aller abgeschlos- ler Enzyme kommt es zur Ablagerung Auch die MPS sind eine heteroge-
senen und noch laufenden klinischen von Glykosaminoglykanen (GAGs; frü- ne Krankheitsgruppe, bei der die kli-
Therapiestudien bei NCL. her Mukopolysaccharide genannt) in nische Ausprägung selbst innerhalb der
verschiedenen Geweben, wovon bei einzelnen MPS-Subtypen variieren kann.
Mukopolysachaccharidosen manchen MPS-Formen auch die Ner- Auch sie werden in der Mehrzahl au-
(MPS) venzellen des Gehirns betroffen sind. tosomal-rezessiv vererbt. Ausnahme bil-
Darüber hinaus sind weitere wichtige det die MPS II (Morbus Hunter), die
Wie die NCL gehören die MPS zu den ly- Organfunktionen eingeschränkt (u. a. einer X-chromosomal-rezessiven Verer-
sosomalen Speicherkrankheiten. Durch Herz, Lunge, Skelettsystem, Sinnesorga- bung unterliegt. In der Regel äußern sich
den Defekt verschiedener lysosoma- ne). die MPS in Form von vergröberten Ge-
236 medizinische genetik 2 · 2018sichtszügen, fortschreitenden Skelettde- wurden etwa 137 Gene identifiziert, wel- Literatur
formitäten, Gelenkkontrakturen, Beteili- che bei Mutationen zu Leukodystrophien
gung von Herz und Lunge, Vergrößerung führen können. Dennoch ist immer noch 1. Kohlschütter A, Schulz A, Bley A et al (2015)
Demenzerkrankungen bei Kindern und Jugendli-
von Leber und Milz sowie fortschreiten- eine signifikante Anzahl an Patienten mit chen. Pädiatr Prax 83:561–570
der psychomotorischer Retardierung. Zu der Diagnose einer „unklaren Leuko- 2. Kohlschütter A, Eichler F (2011) Childhood
neurodegenerativen Symptomen kommt dystrophie“ versehen, die jedoch mithil- leukodystrophies: a clinical perspective. Expert
Rev Neurother 11(10):1485–1496
es vornehmlich bei den Formen MPS III fe der Next Generation Sequenzierung 3. Muenzer J (2011) Overview of the mucopolysac-
und MPS VII sowie den schweren For- sich stetig reduziert. Parallel besteht ei- charidoses. Rheumatology 50:4–12
men der MPS I und MPS II [2]. ne zunehmende Steigerung des Wissens 4. Schulz A, Kohlschütter A, Mink J et al (2013) NCL
diseases – clinical perspectives. Biochim Biophys
Die MPS III wird in die vier Sub- über viele neue Proteine der „weißen Acta 1832(11):1801–1806
typen A–D unterteilt: Bei allen besteht Substanz“, welche wiederum ein verbes- 5. Williams RE, Mole SE (2012) New nomenclature
ein Mangel an einem der vier verschie- sertes Verständnis der Physiologie und and classification scheme for the neuronal ceroid
lipofuscinoses. Neurology 79:183–191
denen Enzyme aus dem Abbauweg des Pathophysiologie dieser Krankheiten be- 6. Cortese A, Tucci A, Piccolo G et al (2014) Novel CLN3
Heparan-Sulfats: Heparan-Sulfamidase wirkt. Vererbt werden die Leukodystro- mutation causing autophagic vacuolar myopathy.
bei MPS IIIA, Alpha-N-Acetylglucosa- phien überwiegend autosomal-rezessiv, Neurology 82(23):2072–2076
7. Schulz A, Simonati A, Laine M, Williams R,
minidase bei MPS IIIB, Alpha-Gluco- eine Ausnahme ist jedoch beispielsweise Kohlschütter A, Nickel M (2015) The DEM-CHILD
saminid:N-Acetyltransferase bei MPS die Adrenoleukodystrophie mit X-chro- NCL Patient Database: a tool for the evaluation of
IIIC und N-Acetylglucosamin-6-sulfat- mosomal-rezessivem Erbgang. therapies in neuronal ceroid lipofuscinoses (NCL).
Eur J Paediatr Neurol 19(Suppl 1):16
Sulfatase bei MPS IIID. Alle vier Formen Die einzelnen Leukodystrophien sind 8. Lebrun AH, Moll-Khosrawi P, Pohl S et al (2011)
werden autosomal-rezessiv vererbt. zwar sehr selten, kommen in der Summe Analysis of potential biomarkers and modifier
Die ebenfalls autosomal-rezessiv ver- jedoch durchaus häufig vor: nach aktuel- genes affecting the clinical course of CLN3 disease.
Mol Med 17:1253–1261
erbte MPS VII wird durch einen Man- ler Berechnung bei 1:7663 Geburten. Ge- 9. LoebelU,SedlacikJ,NickelMetal(2016)Volumetric
gel des lysosomalen Enzyms Beta-D-Glu- naue Aussagen sind allerdings schwierig, description of brain atrophy in neuronal ceroid
kuronidase verursacht. weil die richtige Diagnose nicht immer lipofuscinosis 2: supratentorial gray matter
shows uniform disease progression. AJNR Am J
Die MPS I hat drei Varianten, die gestellt wird. Neuroradiol 37:1938–1943
sich im Schweregrad unterscheiden: Die Während Organe außerhalb des Ner- 10. MoleSE,CotmanSL(2015)Geneticsoftheneuronal
schwerste Form ist das Hurler-Syndrom, vensystems meist nicht betroffen sind, ceroid lipofuscinoses (Batten disease). Biochim
Biophys Acta 1852(10 Pt B):2237–2241
die leichteste das Scheie-Syndrom. Einen führen Leukodystrophien häufig zu fort- 11. Huber RJ, Mathavarajah S (2018) Cln5 is secreted
intermediären Phänotyp hat das Hur- schreitender Spastik sowie progressivem and functions as a glycoside hydrolase in
ler-Scheie-Syndrom. Die Vererbung ist Abbau der kognitiven und sprachlichen dictyostelium. Cell Signal 42:236–248
12. Berkovic SF, Staropoli JF, Carpenter S et al (2016)
autosomal-rezessiv und Ursache der Fähigkeiten. Die Lebenserwartung – be- Diagnosis and misdiagnosis of adult neuronal
unterschiedlichen Phänotypen sind al- sonders bei Kindern – ist meist sehr be- ceroid lipofuscinosis (Kufs disease). Neurology
lelische Mutationen im IDUA-Gen, das grenzt [3]. 87(6):579–584
13. Nickel M, Simonati A, Jacoby D et al (2018) Natural
für die Alpha-Iduronidase kodiert. Die history of late infantile CLN2 disease: quantitative
Mutationen können je nach Schweregrad Korrespondenzadresse prospective assessment of disease characteristics
einen vollständigen (Hurler-Syndrom) and rate of progression in an international cohort
A. Schulz of 140 patients. Lancet Child Adolesc Health.
oder partiellen Enzymdefekt (Scheie- Klinik für Kinder- und Jugendmedizin, https://doi.org/10.1016/S2352-4642(18)30179-2
Syndrom) verursachen. Universitätsklinikum Hamburg-Eppendorf 14. Sun Y, Almomani R, Breedveld GJ et al (2013)
Die MPSII (Mangel der Iduronat-2- Autosomal recessive spinocerebellar ataxia 7
Hamburg, Deutschland
(SCAR7) is caused by variants in TPP1, the gene
Sulfatase) wird als einzige MPS X-chro- anschulz@uke.de involved in classic late-infantile neuronal ceroid
mosomal-rezessiv vererbt. Es erkranken lipofuscinosis2 disease(CLN2 disease). HumMutat
aber nicht nur die hemizygoten Jungen, 34(5):706–713
15. Ku CA, Hull S, Arno G (2017) Detailed clinical
auch wenige betroffene Mädchen mit ei- Einhaltung ethischer Richtlinien phenotype and molecular genetic findings
ner „skewed“ X-Inaktivierung und präfe- in CLN3-associated isolated retinal degeneration.
rentieller Expression des mutierten Allels JAMA Ophthalmol 135(7):749–760
Interessenkonflikt. A. Schulz und M. Nickel geben 16. Ostergaard JR, Rasmussen TB, Molgaard H
wurden beschrieben. an, dass kein Interessenkonflikt besteht. (2011) Cardiac involvement in juvenile neuronal
ceroid lipofuscinosis (Batten disease). Neurology
Dieser Beitrag beinhaltet keine von den Autoren 76(14):1245–1251
Leukodystrophien durchgeführten Studien an Menschen oder Tieren. 17. Lukacs Z, Santavuori P, Keil A et al (2003)
Rapid and simple assay for the determination
Leukodystrophien sind Krankheiten, bei Open Access. Dieser Artikel wird unter der Creative of tripeptidyl peptidase and palmitoyl protein
Commons Namensnennung 4.0 International Lizenz thioesterase activities in dried blood spots. Clin
denen die Myelinentwicklung im zen- (http://creativecommons.org/licenses/by/4.0/deed. Chem 49:509–511
tralen Nervensystem aufgrund eines ge- de) veröffentlicht, welche die Nutzung, Vervielfäl- 18. Schulz A, Ajayi T, Specchio N et al (2018) Sutdy
netischen Defektes gestört ist. Im Zu- tigung, Bearbeitung, Verbreitung und Wiedergabe of intraventricular cerliponase alfa in CLN2
in jeglichem Medium und Format erlaubt, sofern disease. N Engl J Med. https://doi.org/10.1056/
ge dessen kommt es zur Degeneration Sie den/die ursprünglichen Autor(en) und die Quelle NEJMoa1712649
weißer Gehirnsubstanz. Die genetischen ordnungsgemäß nennen, einen Link zur Creative Com-
Ursachen können vielfältig sein; bislang mons Lizenz beifügen und angeben, ob Änderungen
vorgenommen wurden.
medizinische genetik 2 · 2018 237Sie können auch lesen

















































