NEUROKUTANE ERKRANKUNGEN: ALTBEKANNT UND NEU ENTDECKT
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Fortbildung
NEUROKUTANE ERKRANKUNGEN:
ALTBEKANNT UND NEU ENTDECKT
Sandra Tölle
Kurze Einführung Eine inaktivierende Mutation in NF1, einem Tumor Sup-
Die Assoziation von Entwicklungsanomalien von Hirn, pressor Gen, führt zur Fehlfunktion des RAS Inhibi-
Haut und Augen wurde 1932 vom Niederländer Opht- tors Neurofibromin und durch gestörte Regulation von
halmologen Jan van der Hoeve1) unter dem Begriff Pha- Zellwachstum4) zu einer Häufung von benignen und
komatosen zusammengefasst. Er gruppierte darunter malignen Tumoren. NF1 kann fast jedes Organsystem
die Neurofibromatose, die Tuberöse Sklerose und spä- betreffen mit einer breiten Palette an Erscheinungs-
ter die von Hippel-Lindau Erkrankung. Zeitgleich schlu- formen. Die klassische Präsentation ist ein sich mo-
gen Paul Ivan Yakovlev und Riley H. Guthrie auf der an- torisch langsam entwickelndes Kleinkind mit musku-
Sandra Tölle deren Seite des Atlantiks den Begriff Neurokutane Er- lärer Hypotonie, leichter Trichterbrust, relativer Mak-
krankungen vor, geprägt durch ihre Erkenntnisse, dass rozephalie und Café au lait Flecken. Rund 60% der
kongenitale Fehlbildungen des Ektoderms (Haut, Hirn, Kinder leiden im Verlauf unter schulischen Leistungs-
Retina, Auge) wie bei der Neurofibromatose, der tu- problemen und Aufmerksamkeitsdefiziten. Deutlich
https://doi.org/
10.35190/d2021.3.4
berösen Sklerose oder dem Sturge-Weber-Syndrom seltener treten Tumoren des zentralen Nervensys-
klinisch häufig die Gemeinsamkeiten von Hautmani- tems, plexiforme Neurofibrome, Knochenanomalien
festationen, kognitiver Beeinträchtigung und Epilep- (Verbiegung der langen Röhrenknochen, Skoliose)
sie aufwiesen. Sie erkannten auch, dass die Haut ein oder Vaskulopathien (Moyamoya Syndrom) auf. Die
diagnostisches Fenster zum zentralen Nervensystem Diagnose der NF1 kann klinisch gestellt werden (vgl.
ist, was auch heute noch zutrifft2). Tabelle 1 und Abbildung 1), wobei mit Anwendung der
revidierten Diagnosekriterien neu auch der genetische
Beide Begriffe wurden im Verlauf der Jahrzehnte Nachweis einer pathogenen NF1 Variante zählt5).
synonym verwendet. Heute versteht man darunter
eine grosse heterogene Gruppe von Erkrankungen, die Sehbahntumoren (optic pathway glioma)
Haut, Auge und zentrales und peripheres Nervensys- Ca. 15-20% der Kinder mit NF1 entwickeln einen Seh-
tem betreffen, wobei auch andere Organe (z.B. Herz, bahntumor, wobei nur ca. 35% der Sehbahntumoren
Lungen, Nieren, Blutgefässe und Knochen) mitbetei- progredient wachsen, Symptome verursachen und eine
ligt sein können3). Die beschriebenen Erkrankungen Behandlung erfordern. NF1 assoziierte Sehbahngliome
kommen relativ selten vor, aber in der Summe und der werden am häufigsten bei Kindern unter 7 Jahren (Mit-
häufig doch schweren Auswirkungen schon im Kin- telwert 4,5 Jahre) gesehen6). Die Sehbahngliome kön-
desalter bilden sie einen relevanten Anteil im neuro- nen entlang der gesamten Sehbahn auftreten und lo-
pädiatrischen Kollektiv. kalisationsabhängig unterschiedliche Symptome her-
vorrufen. Klinische Zeichen eines Sehbahnglioms
Nach der Erstbeschreibung der «klassischen» Pha- können eine Visusminderung, eine Gesichtsfeldein-
komatosen wie der Neurofibromatose und der Tuberö- schränkung, ein Strabismus, ein relatives Afferenzde-
sen Sklerose Ende des 19. Jahrhunderts, erfolgte Ende fizit, ein Papillenödem oder eine Protrusio bulbi sein.
des 20. Jahrhunderts die genetische Zuordnung und Bei Chiasma- und Hypothalamusbeteiligung können
in den letzten Jahrzehnten dank grosser Fortschritte auch endokrine Auffälligkeiten wie vorzeitige Puber-
in der Molekulargenetik, die Analyse der zellulären Ba- tätszeichen oder ein Wachstumshormonmangel gese-
sis, der intra- und extraneuronalen Signalwege. Dies hen werden bis hin zum sehr seltenen Masseneffekt,
ermöglichte ein besseres Verständnis der zugrunde- dienzephalen Syndrom oder obstruktiven Hydro-
liegenden Pathogenese und letztlich die Entwicklung zephalus. Regelmässige ophthalmologische Kontrol-
von neuen zielgerichteten Therapien. Im Folgenden len sind dementsprechend wichtig und werden halb-
soll am Beispiel von drei neurokutanen Krankheiten jährlich bis jährlich bis zum Alter von sechs Jahren,
auf die neueren Therapiemöglichkeiten eingegangen jährlich bis zum Alter von mindestens zehn Jahren und
werden. alle zwei Jahre bis zum Alter von 18 Jahren empfoh-
len6,7). MRI Untersuchungen als Screening sind nicht
Neurofibromatose Typ 1 (NF1) indiziert, zumal die zufällige Entdeckung eines Seh-
Mit einer Häufigkeit von ca. 1:3000 ist die NF1 die häu- bahntumors bei fehlenden Symptomen nur selten ei-
Korrespondenz: figste neurokutane, autosomal dominant vererbte Er- nen Einfluss hat auf das Management des Patien-
sandra.toelle krankung. In rund 50% der Fälle ist eine Neumutation ten8).
@kispi.uzh.ch verantwortlich für das Tumorprädispositionssyndrom.
14 Vol. 32 | 3-2021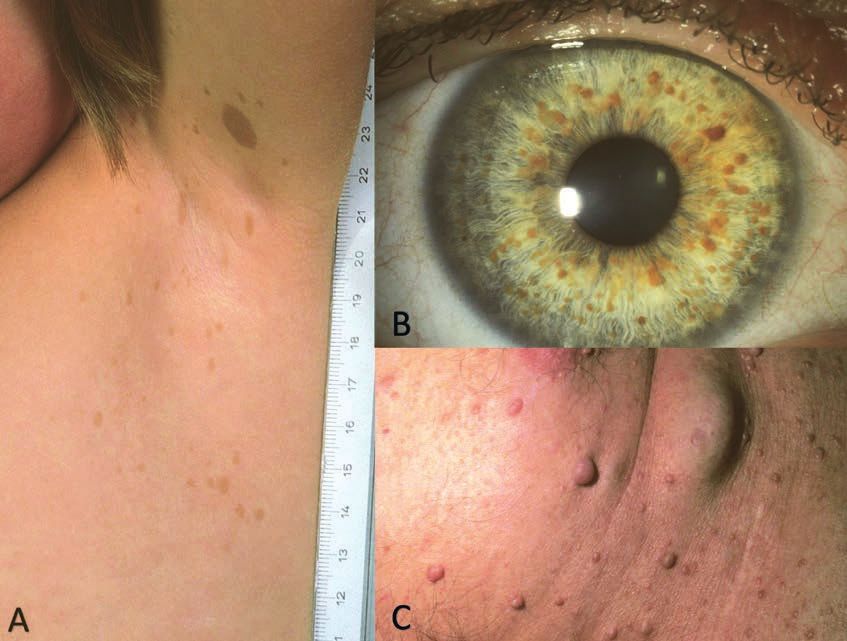
Fortbildung
A Mindestens 2 der folgenden Kriterien müssen erfüllt sein bei einem Kind ohne Elternteil mit NF1
• ≥ 6 Café au lait Flecken ≥ 5 mm präpuberal bzw. ≥ 15 mm postpuberal
• Freckling (vor allem axillär und/oder inguinal)
• ≥ 2 Neurofibrome oder ein plexiformes Neurofibrom
• Sehbahngliom
• ≥ 2 Irishamartome (Lisch-Knötchen) oder ≥ 2 Choroidale Anomalien
• Typische Knochenveränderungen (Sphenoiddysplasie, anterolaterale Verbiegung der Tibia oder
Pseudarthrose eines langen Röhrenknochens)
• Heterozygote pathogene NF1 Variante mit einer Varianten Allelfrequenz von 50% in scheinbar
normalem Gewebe wie Leukozyten
B Mindestens 1 der obigen Kriterien muss erfüllt sein, wenn das Kind einen Elternteil hat mit
NF1 gemäss den unter A gelisteten Kriterien
Tabelle 1: Redividierte diagnostische Kriterien für Neurofibromatose Typ 1
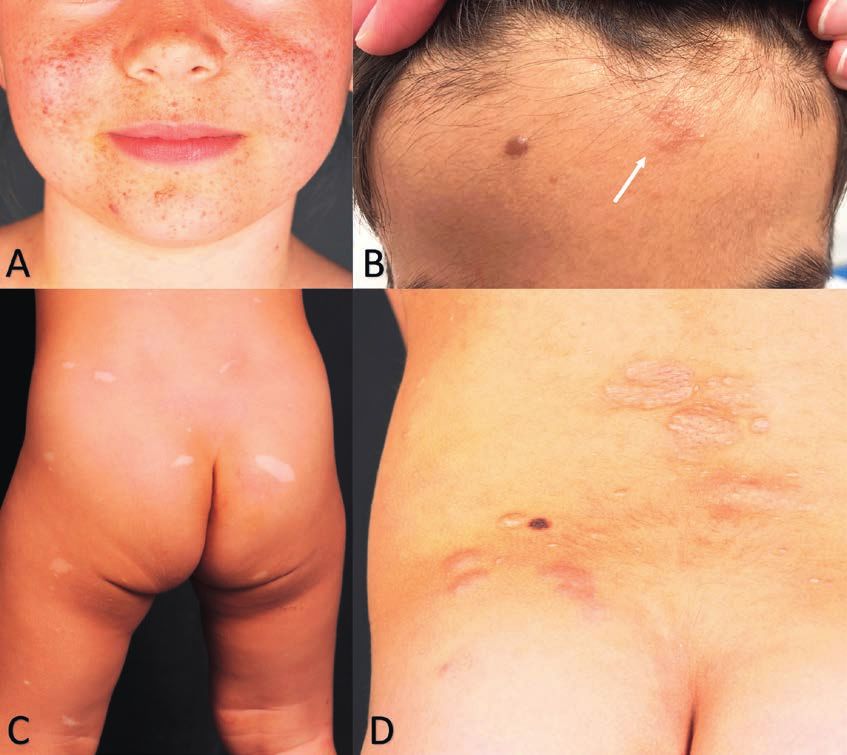
Abbildung 1: Fotos bei Neurofibromatose Typ 1
A Café au lait Fleck und axilläres Freckling
B Lisch Knötchen auf blau pigmentierter Iris
C Kutane Neurofibrome beim Erwachsenen
Behandlung Patienten mit Sehbahntumor und Chemotherapie ge-
Die heutigen Guidelines empfehlen eine Intervention zeigt, dass 32% eine Verbesserung erzielten, 40%
nur, wenn es eine dokumentierte Verschlechterung eine Stabilisierung und 28% eine Verschlechterung.
des Sehens oder einen radiologischen Progress gibt6). Andere Therapiemodalitäten, die üblicherweise bei
Eine Chemotherapie mit Vincristin und Carboplatin Hirntumoren erwogen werden, sind problematisch:
oder nur Vinblastin wird an vielen Kliniken als erster Die Neurochirurgie ist aufgrund der Lokalisation limi-
Behandlungsschritt empfohlen mit der Aussicht auf tiert und die Bestrahlung wird bei Kindern mit NF1 we-
Langzeittumorkontrolle. Bezogen auf die Sehleistung gen dem Risiko von Zweittumoren und Moyamoya Syn-
wurde von Fisher et al.9) in einer Studie mit 115 NF1 drom nicht empfohlen10). Die Anwendung gezielter bio-
Vol. 32 | 3-2021 15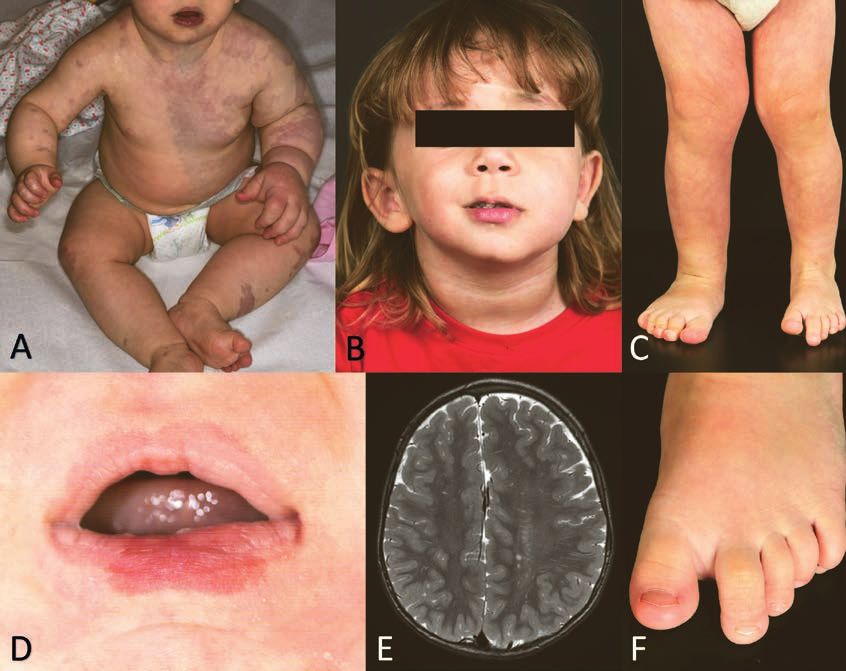
Fortbildung
logischer Therapien bekommt mit der Entschlüsselung (NCI) natural-history study, bei welchen die meisten
der zellulären Pathways einen zunehmenden Stellen- Patienten einen Progress des Tumors zeigten.
wert. Neuere Daten aus präklinischen Modellen haben
die Überaktivierung des MAPK/ERK/MEK Pathway als Tuberöse Sklerose Complex (TSC)
direkte Konsequenz des NF1 assoziierten Funktions- Die TSC ist eine Multisystemerkrankung mit sehr un-
verlustes von Neurofibromin bestätigt11). In der Folge terschiedlicher Ausprägung. Die Häufigkeit der auto-
wurden Studien mit Inhibitoren des Mitogen-activa- somal dominanten Erkrankung wird auf ca. 1:6000
ted protein kinase (MAPK) Weges durchgeführt. Eine -1:10000 geschätzt. Die Mehrheit der Fälle weist eine
kürzlich publizierte Phase-II-Studie mit dem MEK-In- Neumutation auf: Im TSC1 (auf Chromosom 9q34)
hibitor Selumetinib hat bei Patienten mit Rezidiven oder TSC2 (auf Chromosom 16p13) Gen. Die jeweili-
von Low grade Gliomen (inkl. Sehbahntumoren) in 40% gen Genprodukte Hamartin bzw. Tuberin sind wich-
der Fälle ein klares Ansprechen gezeigt12). Nebst Selu- tige Regulatoren des mTORC1-Pathway (mammalian
metinib sind weitere MEK-Inhibitoren in Studien in Er- Target of Rapamycin). Die TSC Mutation führt zur ab-
probung. In einer Phase-II-Studie mit Kindern und NF1 normen Aktivierung dieses mTORC1 Pathway und so
assoziierten low grade Gliomen hat der mTOR-Inhibi- zu unkontrollierter Zellproliferation und Wachstum
tor Everolimus ebenfalls gute Resultate gezeigt13). von Tumoren in vielen Organsystemen. Für die Diag-
nose ausreichend ist der isolierte Nachweis einer pa-
Plexiforme Neurofibrome thogenen Mutation im TSC1 oder TSC2. Im klinischen
Die plexiformen Neurofibrome sind angeborene Tu- Alltag sind es die Haupt- und Nebenmerkmale (Ta-
moren, die sich netzartig entlang peripherer Nerven belle 2), die zur sicheren oder möglichen Diagnose
ausbreiten und keine Wachstumsgrenzen respektie- TSC führen und in der Regel eine Mutationssuche
ren. Sie führen dadurch zu Funktionseinbussen, nach sich ziehen15). Ein negativer Mutationsnachweis
Schmerzen, Entstellung mit verringerter Lebensqua- schliesst eine TSC nicht aus. Heutzutage wird nicht
lität und bei entsprechender Lokalisation auch zur selten bereits pränatal oder neonatal die Verdachts-
Verlegung von Atemwegen. Gross et al.14) haben 2020 diagnose TSC aufgrund multipler kardialer Rhab-
in einer Phase-II-Studie mit 50 Kindern und inoperab- domyome gestellt, die im fetalen Ultraschall entdeckt
len plexiformen Neurofibromen gezeigt, dass mit dem werden. Früher war es meist im Säuglings- und Klein-
MEK-Inhibitor Selumetinib in der Mehrheit der Patien- kindalter der Fall, wenn die Kombination einer Epilep-
ten eine Tumorvolumenreduktion resultierte mit kli- sie (häufig infantile Spasmen) mit weissen Hautfle-
nisch relevantem Benefit z.B. hinsichtlich der Schmer- cken gefunden wurde. Noch heute sind die verschie-
zen. Das Schrumpfen der Tumore und das fehlende denen Hautmanifestationen (Angiofibrome im
Tumorwachstum unter dieser Therapie steht in Kon- Gesichtsbereich, fibröse Stirnplaque oder Shagreen
trast zu den Befunden der National Cancer Institute Patch) pathognomonisch und sollten an die TSC den-
Der Nachweis einer pathogenen TSC1 oder TSC2 Mutation aus nicht betroffenem Gewebe
ist ausreichend für die definitive Diagnosestellung
Hauptkriterien Nebenkriterien
≥3 hypomelatonische Flecken Konfettiflecken der Haut
≥3 Angiofibrome oder Stirnplaque >3 Zahnschmelzdefekte
≥2 unguale Fibrome ≥2 Intraorale Fibrome
Shagreen Patch Unpigmentierter Fleck der Retina
Hamartome der Retina Multiple Nierenzysten
≥3 kortikale Dysplasien (Tuber, Migrationslinien Nichtrenale Hamartome
der weissen Substanz)
Subependymale Gliaknoten
Riesenzellastrozytom
Kardiale Rhabdomyome
Lymphangioleiomyomatose der Lunge (LAM)*
≥2 Angiomyolipome der Niere*
Definitive Diagnose Mögliche Diagnose
2 Hauptkriterien oder 1 Hauptkriterium 1 Hauptkriterium oder ≥ 2 Nebenkriterien
und ≥ 2 Nebenkriterien
* Die Kombination LAM und Angiomyolipome ohne andere Zeichen erfüllt nicht die Anforderungen
für eine definitive Diagnose.
16 Vol. 32 | 3-2021Fortbildung
ken lassen und zur Überweisung an ein spezialisier- ten Therapie geforscht werden, die nicht nur beste-
tes Zentrum führen (Abbildung 2). Ein multidiszipli- hende Symptome lindert, sondern nach Möglichkeit
näres Team ist angesichts der sehr variablen Organ- deren Entstehung verhindern soll. Rapamycin (Siroli-
beteiligung erforderlich. Neben den dermatologischen mus) und das Analogon Everolimus hemmen die
Befunden sind es Manifestationen im ZNS (Epilepsie, mTORC1 Aktivität und führen dadurch zur Kontrolle
Entwicklungsstörung, Riesenzellastrozytom, psychi- von unerwünschten Zellproliferation und Wachstum.
atrische Störungen), Herz (Rhabdomyome), Nieren In Studien, EXIST-119) und EXIST-220), wurde gezeigt,
(Angiomyolipome, Zysten), Lungen (Lymphangiolei- dass mit Everolimus Riesenzellastrozytome des Hirns
omyomatose LAM), Augen (retinale Hamartome), und Angiomyolipome der Nieren kleiner wurden und
Zähne, Knochen und seltener auch in anderen Bau- sich die Lungenfunktion bei LAM stabilisierte. So zeig-
chorganen, die lebenslang kontrolliert und behandelt ten mindestens die Hälfte der Patienten eine Volu-
werden müssen. Ca. 20% der Patienten, meist Kin- menreduktion ihres Riesenzellastrozytoms von > 50%.
der oder Jugendliche, entwickeln ein Riesenzellast- Erwähnenswert ist allerdings die Tatsache, dass nach
rozytom im Bereich des Foramen Monroi, welches bei Absetzen des mTOR-Inhibitors das Tumorwachstum
progredientem Wachstum zu einem obstruktiven Hy- wieder beginnt und darum eine Langzeitbehandlung,
drocephalus führen kann16). Rund 80% der Patienten eventuell sogar lebenslang, nötig ist. Dementspre-
leiden an Angiomyolipomen, die bei zunehmendem chend ist auch eine neurochirurgische Resektion eine
Wachstum mit einer Blutung akut symptomatisch zu diskutierende Option und die Therapieentschei-
werden können und langfristig zur Niereninsuffizienz dung ist in den Gesamtkontext, unter Berücksichti-
führen17). Für die Überwachung und interdisziplinäre gung anderer TSC assoziierten Organmanifestatio-
Behandlung betroffener TSC Patienten existieren in- nen, zu stellen.
ternational breit abgestützte Empfehlungen18).
Ca. 85% der TSC Patienten leiden an einer Epilep-
Behandlung sie, die in der Mehrheit pharmakoresistent ist21). In der
In den letzten Jahren konnte durch das Verständnis EXIST-3 Studie22) wurde an mehrheitlich pädiatrischen
der molekularbiologischen Vorgänge an einer geziel- Patienten gezeigt, dass mit Everolimus auch pharma-
Abbildung 2: Fotos bei Tuberöser Sklerose
A Angiofibrome an Wangen, Nase und Kinn
B Fibröse Stirnplaque links
C Multiple hypomelanotische Flecken
D Shagreen Patch lumbal
Vol. 32 | 3-2021 17Fortbildung
koresistente Epilepsiepatienten bezüglich ihrer Anfälle im Blut gesucht werden. Ein fehlender Mutationsnach-
eine Verbesserung erzielen konnten. Der Anteil Pati- weis schliesst die Diagnose eines PROS nicht aus.
enten mit > 50% Reduktion der Anfallsfrequenz lag
bei 40% in der Gruppe mit hochdosiertem Everolimus Megalencephaly-capillary Malformation
gegenüber 15.1% in der Placebogruppe. Das Präparat Syndrom (MCAP)
wird nebst den bereits genannten Indikationen bei TSC Das MCAP ist ein Vertreter dieser PROS Gruppe, wel-
Patienten auch für weitere TSC assoziierte Krankheits- ches uns im Alltag der neurokutanen Sprechstunde
manifestationen erfolgreich eingesetzt: Schon im Neu- wiederholt begegnet und möglicherweise unterdiag-
geborenenalter bei grossen Rhabdomyomen des Her- nostiziert ist. Die Betroffenen haben einen dispropor-
zens, um eine kardiochirurgische Intervention abzu- tional grossen bis sehr grossen Kopf und kapilläre
wenden und im Kindesalter topisch auf die von Fehlbildungen auf der Haut, gehäuft ein asymmetri-
Angiofibromen betroffene Gesichtshaut. Zusammen- sches Wachstum von Gliedmassen und eine Bindege-
gefasst ist es das Ziel der Behandlung mit einem websauffälligkeit mit Laxizität. Relativ charakteris-
mTOR-Inhibitor, die Grösse und Anzahl der Tumoren tisch ist eine faziale kapilläre Malformation der Ge-
bei TSC Patienten zu verringern oder zumindest zu sichtsmittellinie (Abbildung 3). Ein Teil der Patienten
stabilisieren. Eine neue Multizenterstudie ist geplant, hat zusätzliche ZNS-Fehlbildungen wie Polymikrogy-
um zu untersuchen, ob die prophylaktische Gabe von rien oder eine Hemimegalenzephalie mit dem Risiko
Sirolimus (Behandlungsbeginn vor dem Alter von 4 von Entwicklungsstörungen und Epilepsien. Das sehr
Monaten) eine Reduktion neuropsychologischer Defi- rasche Kopfwachstum in den ersten Lebensjahren mit
zite zur Folge hat. den typischerweise relativ weiten Seitenventrikeln in
der Bildgebung muss sorgfältig, auch mittels MRI, kon-
Erkrankungen durch genetische Mosaike trolliert und interpretiert werden, um einerseits nicht
Verschiedene Entwicklungsstörungen und neuro- unnötig einen Shunt zu implantieren, und andererseits
kutane Erkrankungen sind durch Neumutationen ver- die seltene Komplikation einer Kleinhirnherniation mit
ursacht, die erst nach der Befruchtung (postzygo- Entwicklung eines obstruktiven Hydrozephalus, nicht
tisch) auftreten. Man spricht dann auch von sogenannt zu verpassen. Kopfumfänge von 55-58 cm im Alter
«somatischen» Mutationen. Solche Mutationen haben von zwei Jahren sind keine Seltenheit. Auch beim
zur Folge, dass der Organismus aus genetisch unter- MCAP ist eine somatische PIK3CA Mutation zugrun-
schiedlichen Zellpopulationen besteht24). Obschon deliegend, wobei hier gelegentlich der Mutationsnach-
zahlreiche Mosaikerkrankungen seit vielen Jahren als weis auch im Blut und nicht nur in betroffener Haut
klinische Entitäten bekannt waren, wurde deren zu- gelingt. Der Mutationsnachweis ist wichtig wegen der
grunde liegende genetische Ursache erst in den letz- neuen zielgerichteten Therapiemöglichkeiten. Alpeli-
ten Jahren mit Hilfe der Hochdurchsatzsequenzierung sib (BYL719), ein PIK3CA-Inhibitor, hat in einer Studie
(Next Generation Sequencing, NGS) entdeckt. Ein gu- mit PROS Spektrum Patienten27) zu bemerkenswerten
tes Beispiel dafür ist das sporadisch auftretende Stur- Verbesserungen geführt: Rückgang der kapillären Mal-
ge-Weber-Syndrom, das wie eingangs erwähnt zu den formation und epidermalen Naevi, Verbesserung der
ersten beschriebenen neurokutanen Syndromen ge- Skoliose, Beendigung einer chronischen gastrointes-
hörte, bei dem aber erst 2013 die dafür ursächliche tinalen Blutung. Weitere Resultate aus Studien mit Al-
somatische Mutation im GNAQ Gen in betroffenem pelisib und betroffenen Kindern und Jugendlichen wer-
Gewebe nachgewiesen wurde25). Damit wurde eine seit den bald erwartet.
langem bestehende Hypothese, die der Mosaikerkran-
kung, bestätigt. Für die genetische Beratung der El- Schlussbemerkung
tern verändert der Nachweis einer somatischen Muta- Der Pädiaterin und dem Pädiater wird eine wichtige
tion die Ausgangslage erheblich, besteht doch in die- Rolle zuteil im frühen Erkennen der Verdachtsdiag-
sem Fall bei weiteren Kindern kein erhöhtes Wieder- nose aufgrund der klinischen Symptome und dem Zu-
holungsrisiko für die gleiche Erkrankung. weisen an ein erfahrenes Zentrum. Die Entwicklung
der letzten Jahre mit verbesserten diagnostischen
Vaskuläre Fehlbildungen können assoziiert sein mit und vor allem therapeutischen Möglichkeiten macht
einer breiten Palette von kutanen, neurologischen oder die multidisziplinäre Betreuung dieser Patienten umso
muskuloskelettalen Befunden. Historisch sind zahlrei- wichtiger. Dank: Ich bedanke mich bei Kindern und El-
che Syndrome mit Eigennamen (wie Klippel-Trén- tern für die Erlaubnis zur Publikation der Fotos und
aunay-Syndrom) oder Akronymen (wie CLOVES Syn- bei Lisa Weibel, Martin Theiler und Eugen Boltshau-
drom) und zum Teil überlappenden Phänotypen be- ser für die Mithilfe im Bereitstellen von Bildmaterial.
schrieben worden. Die Entdeckung somatischer
aktivierender Mutationen im PIK3CA Gen hat dann die Für das Literaturverzeichnis verweisen wir auf
zugrundeliegende Ursache verschiedener klinisch de- unsere Online Version des Artikels.
finierter Phänotypen geklärt. Für diese Syndrome mit
der Kombination von vaskulären Fehlbildungen und
segmentalen/fokalen Hypertrophien wurde der neue
Überbegriff PROS für PIK3CA-Related Overgrowth
Spectrum geschaffen26). Der Mutationsnachweis ist
allerdings wegen des Mosaiks nicht einfach und es
muss mit modernen Technologien (NGS) primär in be-
troffenem Gewebe (häufig in Fibroblasten) und nicht
18 Vol. 32 | 3-2021Fortbildung
Abbildung 3: Fotos und MRI Bild bei Megalencephaly-capillary Malformation Syndrom
A Ausgedehnte livide kapilläre Malformation sowie Hypertrophie des linken Armes
B Hypertrophie linke Gesichtshälfte
C Asymmetrisches Körperwachstum mit Hypertrophie rechtes Bein
D Kapilläre Malformation Unterlippe
E Hemimegalenzephalie links passend zur Gesichtsasymmetrie Bild B
F Makrodaktylie der 2. Zehe
Autorin
Dr. med. Sandra Tölle, Abteilung Neuropädiatrie, Universitäts-Kinderspital Zürich - Eleonorenstiftung, Zürich
Die Autorin hat keine finanziellen oder persönlichen Verbindungen im Zusammenhang
mit diesem Beitrag deklariert.
Vol. 32 | 3-2021 19Sie können auch lesen

















































