Zerebrale Anfälle und Epilepsiesyndrome bei Säuglingen - EEG, Semiologie, Ätiologie
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Epilepsie im Säuglingsalter
Zerebrale Anfälle und Epilepsiesyndrome bei
Säuglingen – EEG, Semiologie, Ätiologie
Erlanger EEG-Tage, 14. Juli 2021
Prof. Dr. Regina Trollmann
Leiterin Abt. Neuropädiatrie
und Sozialpädiatrisches Zentrum
Universitätsklinikum Erlangen
Kinder- und Jugendklinik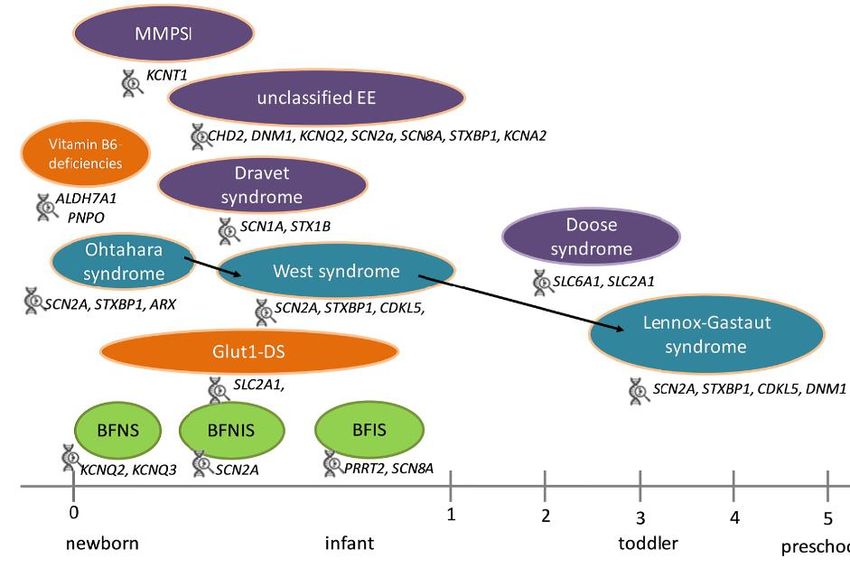
Epilepsien im Säuglingsalter – Manifestation 1.-12. LMo
(AUSWAHL)
Neue ILAE Klassifikation 2017
Fokale Epilepsien und Syndrome
Ätiologie ➢Symptomatisch fokal
KOMORBIDITÄTEN
Strukturell
➢BNS-Epilepsie (multifokal)
Epilepsietyp
Genetisch
Fokal generalisie Kombiniert un- ➢Epilepsie der frühen Kindheit mit
rt generalisiert & bekannt
Infektiös
fokal migratorischen fokalen Anfällen
Metabolisch (EIMFS)
Immuno-
logisch ➢ Watanabe Syndrom (4-8 Mo)
Epilepsiesyndrom Unbekannt ➢Atypische benigne fokale Epilepsien,
CSWS-Syndrom (ab 8 Mo)
Scheffer et al. 2017
Epilepsien im Säuglingsalter – Manifestation 1-12 Mo
(AUSWAHL)
Generalisierte genetische
Epilepsien und Syndrome
➢ Benigne myoklonische Epilepsie
des Kleinkindalters (4 Mo-3 Jahr)
➢ Schwere myoklonische Epilepsie
des Kindesalters (SMEI) (Dravet
Syndrom)
➢ Myoklonisch atonische Epilepsie
Weber et al. Expert Review of Molecular Diagnostics 2017
(Doose)
➢ „Epileptische Enzephalopathien“BNS-Epilepsie, West-Syndrom, infantile Spasmen
Ehem. Zwillings-FG, 29. SSW; Alter 7 Mo Altersabhängige Epilepsie des
Säuglingsalters
(Gipfel 4.-8. LMo; M>W)
Inzidenz 3-4.5/10 000
Lebendgeb.
TRIAS:
BNS-Anfälle (Blitz-Nick-
Salaam)
Hypsarrhythmie
Entwicklungsstillstand/Regr
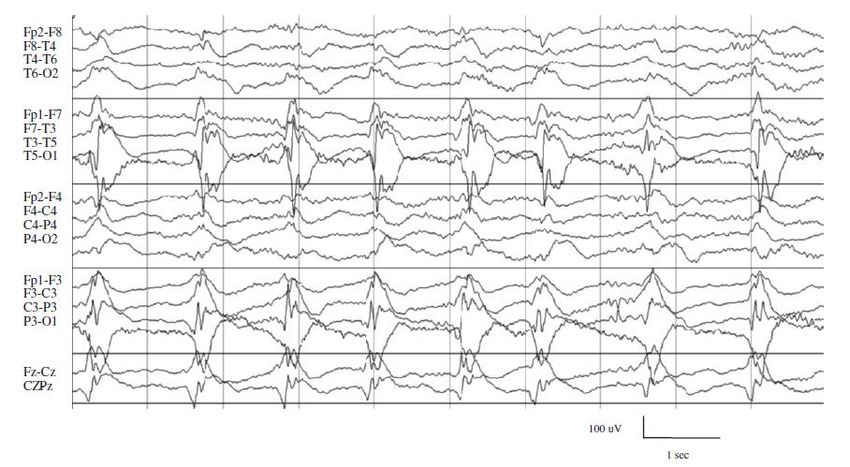
essionBNS-Epilepsie Hypsarrhythmie (hypselos =hoch; arrhythmia)
EEG
7 Mo, Im Schlaf ◼ Generalisierte langsame Wellen wechselnder,
hoher Amplitude (>100 – 500µV)
◼ in wechselnder Lokalisation eingelagerten spikes
und sharp waves variabler
Amplitude/Frequenz/Morphologie
▪ Fokale ETP (30% d. F.)BNS-Epilepsie
Semiologie
◼ Myoklonische (Blitz-) Anfälle ◼ Häufig in Clustern
▪ Intervalle 5-30 sec
▪ Bilateral symmetrisch>asymmetr.
▪ kurz nach dem Erwachen
▪ Beuge-> Streckmuster
◼ Assoziierte Phänomene
▪ Schreien im Intervall/nach den
◼ Nickanfälle Anfällen
▪ Bulbusdeviation, Nystagmus
▪ tonisch
◼ Entwicklungsstillstand/-regression
◼ Vorausgehende fokale/multifokale
◼ Tonische Beugekrämpfe (Salaam) Anfälle in 20-30% d. F.
▪ Bilateral symmetrischBNS-Epilepsie
Ätiologie als wichtigster prognostischer Faktor
>90% symptomatisch, > 55% prä-/perinatale Ursache
Strukturell
➢ Läsionen: Hypoxisch-ischämisch, Stroke, postinfektiös
➢ Kortikale Malformationen
➢ Neurokutane Erkrankungen (TSC, Hypomelanosis Ito, Sturge-Weber)
Metabolisch
➢ Nicht-ketotische Hyperglycinämie, Pyridoxin-, Folinsäure-Defizienz, Mitochondriopathien,
Molybdän-Cofaktor-Mangel....
Genetisch
➢ Chromosomenanomalien (Trisomie 21,…)
➢ CDKL5, SLC25A22, SPTAN1, PLCB1, ST3GAL3, GRIN2B, ...
➢ Keine systematischen Daten
Sumanasena et al. DMCN 2019; Paciorkowski et al. Pediatr Neurol 2011; Song et al. Clin Neuropharmacol
2017Tuberöse Sklerose Komplex (AD, TSC1/TSC2-Genmutationen) TSC und Epilepsie: > 80% der Patienten >70% Manifestation im Säuglingsalter Pharmakoresistenz 65% Indikatoren für Pharmakoresistenz/Entwicklungsstörung o BNS-Anfälle (kognitive Defizite 76%) o Komplex-fokale Anfälle, refraktär im Säuglingsalter o Initiale EEG-Befunde, Tubera/Noduli-Last cMRT o TSC2-Mutation
Version 2.0 (2014-2019)
ACTH 15IU/m2/d für 2 Wo
30IU/m2/d für 2 Wo (Reduktion über 18 Wo)
Steroide Prednisolon, Prednison 2-5 mg/kg/d (>4 Wo)
Vigabatrin (1. Wahl bei Tuberöser Sklerose)
75-150 mg/kg/d (6 Mo)
Bei Therapieversagen Pyridoxin, Valproat, Benzodiazepine, Topiramat, Zonisamid
Steroide + Vigabatrin als First line
Refraktärer Verlauf und
Zu Steroiden und Vigabatrin vergleichbare Effizienz schwere mentale
Topiramat
Retardierung: > 70 % d.F.
Levetiracetam
Zonisamid
Valproat
Sumanasena et al. DMCN 2019, Song et al. Clin
Ketogene Diät Neuropharmacol 2017; Iyer & Appleton, Pediatric Drugs 2016Epilepsie der frühen Kindheit mit migratorischen fokalen
Anfällen (EIMFS) Manifestation ab 3. LMo
Semiologie
Autonome Sy 43 %
fokal motorisch 64 %
(+okulär/fazial)
Alternierende
Hemi-Kloni 59 %
Bilat. ton-klon. 17 %
Spasmen 7%
Kuchenbuch et al. Epilepsia 2019; Auvin et al. Neurobiol Dis
2016; McTague et al. Brain 2013Epilepsie der frühen Kindheit mit migratorischen fokalen
Anfällen (EIMFS) Manifestation ab 3. LMo
Entwicklung Normal vor Epilepsiebeginn 100%; Mikrozephalie 75%,
Dystonie/Choreoathetose 23%, Regression 100%
Häufigste Ätiologie
✓ KCNT1 (Na-aktivierter Kaliumkanal-Subtyp; Gain-of-function), bis zu
40% d. F.
▪ Chinidin (limitierte Datenlage)
Mikati et al. 2015; Fukuoka et al. 2017; Zhou et al. Genes Brain Behav 2018; Ambrosio et
al. Ann Neurol 2018Dravet-Syndrom - Severe myoclonic epilepsy in infancy (SMEI)
Klinik
Manifestation 3.-9. Mo, m>w (2:1)
Beginn häufig mit “Fieberkrämpfen“
✓ ! frühe Rezidive, prolongiert, Hemi-GM, Myoklonien im Verlauf
Status epilepticus häufig!
✓ Konvulsiv (häufig Fieber-induziert, prolongiert)
✓ Subklinisch (±irreguläre polytope Myoklonien)
✓ Ab 2.-3. Lj Störungen der Entwicklung
✓ Sprache, Wahrnehmung, Ataxie
✓ Photosensitivität
✓ FA positiv für Fieberkrämpfe, gen. Epilepsien (31%)
✓ SCN1A-Mutationen
Meng et al. Hum Mutat 2015; Djemie et al. Mol Genet Genomic Med 2016; Carvill et al. Neurology 2014, Balestrini et al. Acta Neurol Scand.
2017; Ceulemann et al. Epilepsia 2016, Wilmshurst et al. Epilepsia 2015Dravet-Syndrom Behandlungsoptionen limitiert Günstig VPA, K-Bromid, Stiripentol, BZD (Clobazam) Unklar TPM, LEV, ketogene Diät Kontraindiziert CBZ, LTG, PHT, LCM Orphan Drug Cannabidiol, Fenfluramin Prognose ✓ 50% kognitive Defizite ✓ Ataxie, spastische Bewegungsstörungen ✓ Verhaltensstörungen, Psychosen ✓ Letalität 9-16% (SUDEP)
Myoklonisch atonische Epilepsie
(Doose Syndrom)
➢ Beginn im 1. LJ bei 24% (GTKA) – 5. LJ
➢ Myoklonische, atonische Anfälle, Atypische Absencen
➢ Status in 40% (Absencen, myoklonisch-atonische Anfälle)
➢ MRT normal
IQ normal 58% - leichte kognitive Defizite ca. 20%
Schwere kognitive Defizite 22%
Refraktäre Verläufe: DD GLUT1-Defekt (SCL2A1-Mutation),
SLC6A1-Mutationen (GABA-Transporter) Weber et al. 2017
Keine genauen Daten !GLUT1-Defekt – klassische Form SLC2A1-Mutationen in >80% d.F. Refraktäre, früh manifeste Epilepsie (
Epileptische Enzephalopathien
Leitsymptome für eine erweiterte metabolische Diagnostik
o Frühe Manifestation + Pharmakoresistenz
o Progressive myoklonische Anfälle
o Enzephalopathie
✓ Akute kristenhafte Verschlechterung
✓ Entwicklungsregression
✓ MRT-Befunde
✓ Mikro-/Makrozephalie
✓ Ophthalmologische Befunde
✓ Organomegalie
✓ Positive FA, Konsanguine ElternFrühkindliche mitochondriale Enzephalomyopathien
◼ Hypotonie +/- Schwäche Variable Laktat
◼ Enzephalopathie MRT ZNS
◼ Kardiomyopathie Genetik: Molekulargen. Defekte häufig
unbekannt (mtDNA-kodiert: COX I, IV> II, III;
◼ Tubulopathie nDNA-kodiert: SURF-1), POLG1
◼ (Ptose/Ophthalmoplegie) Finsterer J. J Neurol Sci 2019; Salar et al. Epilepsia Open
2018; DiMauro S et al. Adv Exp Med Biol 2012Mitochondriale hepatozerebrale Erkrankung
Morbus Alpers (POLG1 Mutationen, aut-rez)
Refraktärer CSE als
Erstsymptom im
Säuglings-/Kleinki-Alter
▪ Entwicklungsregression
▪ Ataxie
▪ Refraktäre Epilepsie,
myoklonisch, Epilepsia
partialis continua
▪ Leberdysfunktion
(biochemisch)
▪ MRT:
RHADS: fokale okzipitale, rhythmische Delta
mit Überlagerung von Polyspikes
kortikale/subkortikale T2
Hyperintensitäten
McCoy et al. Eur J Pediatr Neurol 2011
Hikmat et al. Genet Med. 2017Tyrosinhydroxylase-Mangel
Missens-Mutationen im Tyrosinhydroxylase-Gen (11p15.5), AR
Störung der Biosynthese der Diagnose
Katecholamine Dopamin, NA, A ▪ Klinik/Okulogyre Krisen, selten
zerebrale Anfälle
▪ Neurotransmitter im Liquor
▪ Molekulargenetik
Therapie
▪ L-Dopa/Carbidopa
Prognose
▪ Hohes Risiko motorischer und
mentaler Retardierung
Brennenstuhl et al. Neuropediatrics 2018Diskussion Semiologie und Ätiologie von Epilepsien im Säuglingsalter ✓ EEG zur Diagnosesicherung epileptischer Aktivität beim Säugling essentiell - häufig atypische Semiologie ✓ Prognostisch entscheidend: Ätiologie/Komorbiditäten ✓ Ätiologische Heterogenität & phänotypische Variabilität ✓ Genetische Diagnostik frühzeitig
Sie können auch lesen

















































