Mitochondriale Erkrankungen - DGM-Handbuch Ein Patientenratgeber - Herausgegeben von der DGM Deutsche Gesellschaft für Muskelkranke e.V - mitoNET
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten

DGM-Handbuch
Mitochondriale Erkrankungen
Ein Patientenratgeber
Herausgegeben von der DGM · Deutsche Gesellschaft für Muskelkranke e.V.
2. Auflage · November 2017
1
DGM-Handbuch
Mitochondriale Erkrankungen
Ein Patientenratgeber
Mit freundlicher Unterstützung
der AOK Baden-Württemberg
Hauptverwaltung
Autoren:
Dr. med. Viktoria Bau, Dresden
Dr. med. Matthias Boentert, Münster
Dr. med. Boriana Büchner, München
Prof. Dr. med. Marcus Deschauer, München
Prof. Dr. med. Peter Freisinger, Reutlingen
Dipl.-Psych. Carsten Gamroth,
Psychologischer Psychotherapeut, Lübeck
Impressum Prof. Dr. Werner Klingler, Ulm
Prof. Dr. med. Thomas Klopstock, München
Prof. Dr. Dr. h.c. Frank Lehmann-Horn, Ulm
Dr. Holger Prokisch, München
DGM
Deutsche Gesellschaft
Dr. Carsten Schröter, Bad Sooden-Allendorf
Sandra Sittinger, Halle
Prof. Dr. med. Wolfgang Sperl, Salzburg
für Muskelkranke e.V.
PD Dr. med. Dr. Sakia B. Wortmann,
München und Salzburg
Diagnosegruppe
Mitochondriale Erkrankungen Prof. Dr. med. Ali Yilmaz, Münster
Prof. Dr. med. Peter Young, Münster
Im Moos 4
79112 Freiburg
Telefon: 0 76 65 / 94 47-0 Mito-Diagnosegruppe der DGM:
Telefax: 0 76 65 / 94 47-20
E-Mail: info@dgm.org Karin Brosius, München
Internet: www.dgm.org Claus-Peter Eisenhardt, Lauffen/Neckar
2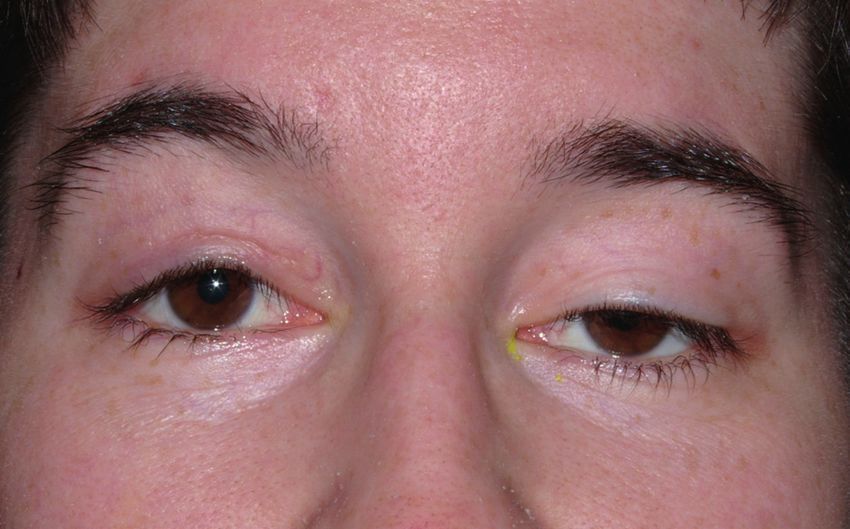
INHALT
Vorwort.......................................................... 4 Herzbeteiligungen........................................ 27
Claus-Peter Eisenhardt, Lauffen/Neckar Prof. Dr. med. Ali Yilmaz, Münster
Karin Brosius, München
Physiotherapie und Rehabilitation.............. 30
Wissenswertes über die................................ 5 Dr. Carsten Schröter, Bad Sooden-Allendorf
Mitochondrialen Erkrankungen
Prof. Dr. med. Marcus Deschauer, München Anästhesie................................................... 33
bei mitochondrialen Erkrankungen
Mitochondriale Erkrankungen....................... 7 Prof. Dr. Werner Klingler, Ulm
mit Auftreten im Erwachsenenalter Prof. Dr. Dr. h.c. Frank Lehmann-Horn, Ulm
Prof. Dr. med. Marcus Deschauer, München
Atmungsprobleme....................................... 36
Mitochondriale Erkrankungen....................... 9 bei Patienten mit mitochondrialen
mit Auftreten im Kindesalter Erkrankungen und deren
Prof. Dr. med. Peter Freisinger, Reutlingen Behandlungsmöglichkeiten
Prof. Dr. med. Wolfgang Sperl, Salzburg Dr. med. Matthias Boentert, Münster
Prof. Dr. med. Peter Young, Münster
Diagnostik.................................................... 12
PD Dr. med. Dr. Sakia B. Wortmann, Fehldiagnosen.............................................. 39
München und Salzburg Prof. Dr. med. Thomas Klopstock, München
Dr. Holger Prokisch, München
Das Deutsche Netzwerk............................... 38
Übersicht...................................................... 15 für mitochondriale Erkrankungen (mitoNET)
zu den Therapiemöglichkeiten Prof. Dr. Thomas Klopstock, München
Prof. Dr. med. Marcus Deschauer, München Dr. med. Boriana Büchner, München
Prof. Dr. med. Peter Freisinger, Reutlingen
Prof. Dr. med. Wolfgang Sperl, Salzburg Spenden zu Gunsten.................................... 43
der Mito-Diagnosegruppe/-Forschung
Mitochondriale Erkrankungen..................... 17 Claus-Peter Eisenhardt, Lauffen
und Lebensqualität – Karin Brosius, München
Langfristige Krankheitsfolgen
frühzeitig beachten und behandeln! Was tut die DGM für Muskelkranke............ 44
Dipl.-Psych. Carsten Gamroth,
Psychologischer Psychotherapeut, Lübeck
Beitrittserklärung......................................... 45
Augenbeteiligungen..................................... 22
Dr. med. Viktoria Bau, Dresden
Schluckstörungen......................................... 25
Prof. Dr. med. Marcus Deschauer, München
Sandra Sittinger, Halle
3
Vorwort
Bei dem Ihnen hier vorliegenden Patientenrat- – auch unter dem Namen Mitochondriopatien
geber dem „DGM-Handbuch Mitochondriale bekannt – sind in den letzten Jahren bahnbre-
Erkrankungen“ handelt es sich um eine neue chende Entwicklungen zu verzeichnen. Engagier-
Informationsbroschüre, die den aktuellen Wis- ten Wissenschaftlern gelang es mittels neuer
sensstand auf diesem Gebiet wiederspiegelt. Seit genetischer Methoden fast 300 Gene zu identi-
der Gründung unserer Selbsthilfegruppe im Jahr fizieren, welche mit mitochondrialen Defekten
2006 hatten wir bereits zwei Broschüren her- und Dysfunktionen assoziiert werden. Trotz der
ausgegeben. Die Entwicklung vom ursprünglich verstärkten Forschungstätigkeit in den letzten
achtseitigen Faltblatt hin zu diesem umfangrei- Jahren und dem Zuwachs an Kenntnissen in der
chen Handbuch, nun in der 2. aktualisierten Auf- Grundlagenforschung zeichnet sich leider immer
lage, zeugt eindrucksvoll von einem gestiegenen noch kein fundamentaler Durchbruch in Richtung
Interesse an der wissenschaftlichen Forschung Heilung der mitochondrialen Erkrankungen ab.
zu den mitochondrialen Erkrankungen, das Umso wichtiger bleibt, neben der medizinischen
nicht zuletzt auf die Entstehung des Deutschen Versorgung, die „Selbsthilfe“, d.h. der Umgang
Netzwerkes für Mitochondriale Erkrankungen mit der Krankheit und den damit verbundenen
(mitoNET) zurückzuführen ist. Die Gründung Behinderungen im Alltag. Dieser Säule schenken
unserer Selbsthilfegruppe und die Etablierung wir weiterhin viel Aufmerksamkeit.
des wissenschaftlichen Forschungsverbundes
mitoNET erfolgten ungefähr zur gleichen Zeit und Wir laden alle Betroffenen ein, sich mit uns in
wir standen als Patientenvertreter von der ersten der Selbsthilfe zu engagieren und so eigene
Stunde an im Dialog mit den führenden Wissen- Wege zur Steigerung der Lebensqualität im Aus-
schaftlern und Klinikern auf diesem Gebiet. Von tausch mit anderen Betroffenen zu finden. Aber
diesem Dialog profitierten beide Seiten. Wir auch die notwendige Lobbyarbeit zu betreiben,
Patientenvertreter lernten viel über unsere um das Bewusstsein für diese seltene Krankheit
Krankheit und den neuesten Stand der For- und ihre gravierenden Folgen bei den politischen
schung und konnten dieses Wissen in unzähligen und gesellschaftlichen Entscheidungsträgern zu
Beratungsgesprächen und auf Fachtagen an erhöhen.
Betroffene weitergeben, die mit dieser Diagnose Kontaktadressen sind über die Geschäftsstelle
oder Verdachtsdiagnose konfrontiert wurden. der DGM in Freiburg und über die Internetseiten
Umgekehrt konnten wir die Mediziner über Sym- www.dgm.org bzw. www.mito-erkrankungen.de
ptome der Patienten informieren (z. B. Schluck- erhältlich.
beschwerden, unspezifische Schmerzen), deren
häufiges Auftreten den Ärzten vorher gar nicht Ihr Vorsitzender und Ihre stellvertretende Vorsit-
bewusst war und nun bei der Anamnese wesent- zende der Mito-Diagnosegruppe in der DGM e.V.
lich mehr Beachtung findet.
Claus-Peter Eisenhardt und Karin Brosius
Dieses Handbuch ist Ausdruck der guten Zusam-
menarbeit zwischen der Mito-Diagnosegruppe
und der Wissenschaft und greift neben dem
aktuellen Überblick über die Grundlagen der
mitochondrialen Erkrankungen nun auch die Kontakte zur Diagnosegruppe Mitochondriale
häufigsten Fragen von Patienten auf. Zudem Erkrankungen in der DGM e.V.
widmet es sich auch den verschiedenen Thera- Die Ansprechpartner der Mito-Diagnosegruppe
piemöglichkeiten wesentlich ausführlicher als in finden Sie aktuell auf der Homepage
der früheren Auflage. Wir hoffen, dass wir damit www.mito-erkrankungen.de oder www.dgm.org
dem Informationsbedürfnis der Betroffenen über und über die DGM Bundesgeschäftsstelle unter
diese seltene, so komplexe und heterogene Er- Tel. 0 76 65 / 94 47-0
krankung noch besser Rechnung tragen können.
In der Diagnostik mitochondrialer Erkrankungen
4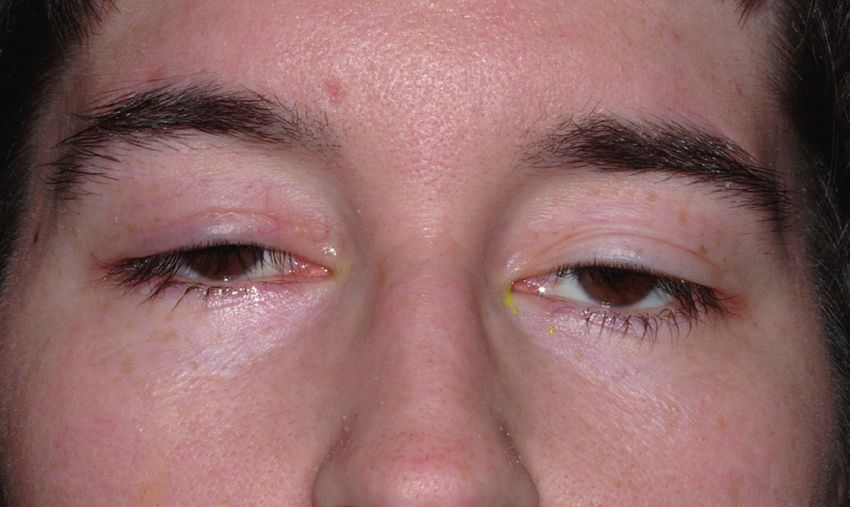
Wissenswertes über die Mitochondrialen Erkrankungen
Prof. Dr. med. Marcus Deschauer, München kommen. Aber auch andere Zellen und Gewebe
können betroffen sein, so z. B. das Nervensys-
tem, das Auge und das Innenohr. Störungen des
Was sind Mitochondrien? Magen-Darm-Traktes, der Leber des Herzens
Mitochondrien sind Organellen (Strukturen der oder der Bauchspeicheldrüse kommen ebenfalls
Körperzellen mit bestimmter Funktion), die für vor.
die Energiegewinnung der Zellen verantwortlich
sind. Man kann sie als „Kraftwerke“ der Zellen Bei Erkrankungen der Mitochondrien handelt es
bezeichnen. In der sog. Zellatmung findet die sich daher oft um sog. Multisystemerkrankun-
„Verbrennung“ von Sauerstoff statt. Dazu gibt es gen, bei denen verschiedene Organe erkranken.
die sog. „Atmungskette“ an der inneren Memb- Eine Mitochondrien-Erkrankung kann sowohl im
ran der Mitochondrien. Darüber hinaus haben Kindesalter als auch im Erwachsenenalter auf-
die Mitochondrien aber noch weitere Funkti- treten, die klinische Ausprägung kann im Verlauf
onen, so sind sie z. B. auch für den Abbau der wechseln.
Fettsäuren verantwortlich.
Häufig berichten Patienten mit mitochondrialen
Allgemeines – Mitochondriopathien sind sehr Myopathien über eine belastungsabhängige
vielgestaltige Erkrankungen Muskelschwäche, allgemeine Erschöpfung und
Mitochondriopathien sind Erkrankungen, bei Belastungsintoleranz. Aber auch eine andauern-
denen ein Defekt in den Mitochondrien vorliegt, de Muskelschwäche ist möglich. Wegweisend
der in der Regel eine genetische Ursache hat. für eine mitochondriale Myopathie ist, dass die
Funktionsstörungen der Mitochondrien betreffen Augenmuskeln betroffen sind. Bei manchen Pati-
insbesondere die Muskelzellen, da diese einen enten kann eine Herzmuskelschwäche auftreten,
hohen Energiebedarf aufweisen. Es kann zu einer aber auch Störungen der Nervenleitung im Herz
mitochondrialen Myopathie (Muskelerkrankung) sind möglich, was zu Herzrhythmusstörungen
(Abb. 1).
5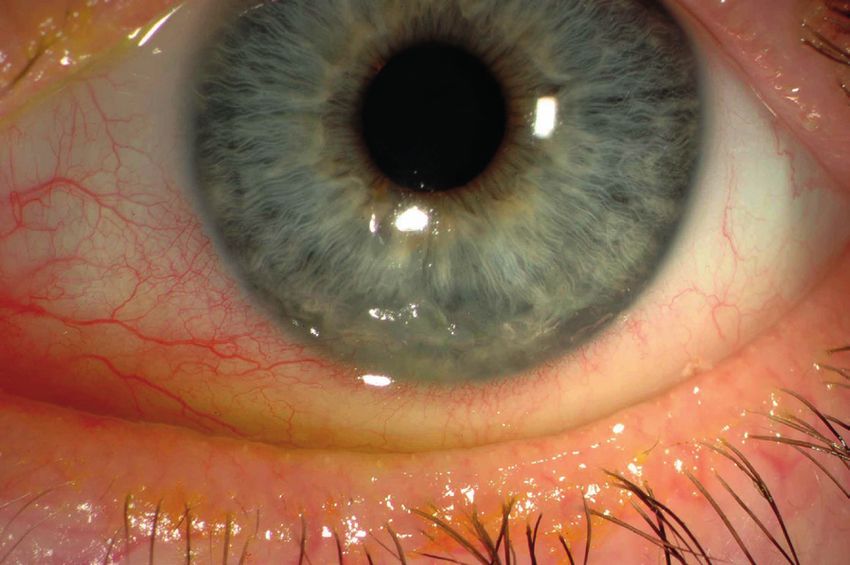
führen kann. Ist das Gehirn betroffen, können selbst gebildet werden können. Viele Eiweißstof-
z. B. epileptische Anfälle, geistige Behinderungen fe werden aber von der Kern-DNA gebildet und
oder schlaganfallähnliche Beschwerden auftre- in die Mitochondrien hinein transportiert. Liegt
ten. Die Nerven von Armen und Beinen kön- der Fehler in der Erbsubstanz der Mitochondri-
nen ebenfalls betroffen sein. Am Auge können en, so wird die Erkrankung typischerweise von
besonders der Sehnerv und die Netzhaut erkran- der Mutter auf die Kinder übertragen (maternale
ken. Außerdem sind die Betroffenen häufig mit Vererbung), da die mitochondriale DNA nur von
Schwerhörigkeit und Diabetes belastet. der Mutter an die Kinder weiter gegeben wird.
Dies erklärt sich dadurch, dass die Eizellen im
Typische Kombinationen von Symptomen können Vergleich zu den Spermien tausendfach mehr
den Arzt zur Diagnose der Mitochondriopathie Mitochondrien enthalten.
führen. Man spricht dann von charakteristischen
Syndromen. Einige dieser Syndrome werden Bei Veränderungen der mitochondrialen DNA
später im Einzelnen vorgestellt. Allerdings wei- ist meist nur ein Teil der DNA-Moleküle betrof-
sen viele Betroffene nicht das volle Bild dieser fen, so dass in einer Gewebeprobe normale
Syndrome auf und es gibt manchmal auch eine mitochondriale DNA neben mutierter mitochon-
Überlappung zweier Syndrome. drialer DNA gefunden werden kann (Hetero-
Mitochondriopathien gelten als seltene Erkran- plasmie). Dabei gibt es eine gewisse Beziehung
kungen. Einzelne Syndrome sind sogar sehr zwischen dem Anteil der veränderten DNA und
selten. dem Schweregrad der Erkrankung. Leider ist bei
Mutationen der mitochondrialen DNA eine gene-
Genetische Grundlage tische Beratung von Familien und insbesondere
Mitochondriopathien liegen in der Regel Feh- Pränataldiagnose sehr schwierig, da der Hete-
ler (Mutationen) in der Erbsubstanz zugrunde, roplasmiegrad beim Kind in den verschiedenen
wobei nur manchmal eine Erbkrankheit in der Geweben sehr unterschiedlich sein kann.
Familie offensichtlich ist. Allerdings findet man
nicht bei allen Patienten den Fehler in der Liegt der Fehler aber in der Kern-DNA, so kön-
Erbsubstanz, besonders bei Kindern bleibt der nen auch Väter die Erkrankung weiter geben
Gendefekt manchmal unbekannt. Neue Metho- (autosomal dominanter Erbgang). Viel häufiger
den in der Molekulargenetik („Next-Generation- ist bei Mitochondriopathien durch Fehler in der
Sequenzierung“), die seit wenigen Jahren zu Kern-DNA aber ein sog. autosomal rezessiver
Einsatz kommen, erleichtern die Suche erheblich. Erbgang, wo in der Regel die Erkrankung auf eine
Generation beschränkt bleibt und normalerweise
Die Mitochondrien haben eine eigene Erbsub- nur Geschwister ein Erkrankungsrisiko haben.
stanz (mitochondriale DNA), die zusätzlich zur Die Vererbbarkeit der einzelnen Erkrankungen
Erbsubstanz im Zellkern in der Zelle vorliegt, wird bei der Beschreibung der Krankheitsbilder
so dass einige Eiweißstoffe im Mitochondrium erläutert.
6
Mitochondriale Erkrankungen mit Auftreten im Erwachsenenalter
Erkrankungen durch Störung der Zellatmung der Jugend auf und liegen Netzhautverände-
(Defekte der Atmungskette) rungen am Auge und Nervenleitungsstörungen
im Herz vor, so spricht man vom Kearns-Sayre-
Syndrom.
Krankheiten mit Auftreten typischerweise
im Jugend- oder Erwachsenenalter Wie es der Name CPEO bereits sagt, handelt es
sich um eine chronisch fortschreitende Erkran-
Prof. Dr. med. Marcus Deschauer, München kung, wobei das Fortschreiten in der Regel sehr
langsam ist. Daher wird die Muskelschwäche in
den Beinen in der Regel selten so schlimm, dass
Chronisch progressive externe Patienten nicht mehr laufen können. An den
Ophthalomoplegie (CPEO) Augenmuskeln kann hingegen im Verlauf eine
Diese Erkrankung ist durch eine langsam zuneh- vollständige Lähmung auftreten, die hinsichtlich
mende (chronisch progressive) Lähmung (Plegie) der Augenbeweglichkeit jedoch wenig Beschwer-
der äußeren (externen) Augen (Ophthalmo)- den macht, hinsichtlich des Hängens der Lider
Muskeln charakterisiert, was zur Bewegungs- aber schon. Die multisystemischen Beschwerden
einschränkung der Augen und zum Hängen der über die Muskeln hinaus können auch im Verlauf
Lider (Ptose) führt. Die Patienten bemerken die der Erkrankung hinzutreten, so dass regelmäßige
einschränkte Augenbeweglichkeit jedoch häufig Kontrolluntersuchungen dahingehend zu emp-
nicht selbst, da selten Doppeltsehen auftritt. fehlen sind.
Vielmehr fällt das Herabhängen der Lider auf, Die CPEO ist eine sehr häufige Mitochondriopa-
das nicht nur ein kosmetisches Problem darstellt, thie, die sehr unterschiedlich vererbt wird. Meist
sondern auch zu einer Sehbeeinträchtigung nach weisen die Patienten sog. einzelne (singuläre)
oben führt, so dass die Patienten den Kopf nach Verkürzungen (Deletionen) der mitochondrialen
hinten neigen. Bei Fortschreiten kann die Pupille Erbsubstanz auf, die nur sehr selten von betrof-
ganz verdeckt werden, so dass der Patient nur fenen Müttern an die Kinder vererbt werden (ca.
etwas sieht, wenn er das Lid mit dem Finger 4%). Andere Patienten haben sog. mehrfache
anhebt. Zusätzlich zur Augenmuskellähmung (multiple) Verkürzungen der mitochondrialen
weisen viele, aber nicht alle Patienten, weitere DNA. Diese Veränderungen sind die Folge von
Beschwerden auf, man spricht dann von „CPEO Defekten in Genen der Kern-DNA, die autosomal
plus“. dominant oder rezessiv vererbt werden. Es sind
bislang 15 Gene bekannt (am häufigsten treten
Dazu gehört insbesondere eine belastungsab- Defekte im POLG-Gen auf). Selten finden sich
hängige Muskelschwäche, die besonders die sog. Punktmutationen der mitochondrialen Erb-
rumpfnahen Muskeln von Armen und Beinen substanz, die von der Mutter vererbt werden.
(Schultergürtel/Oberarme bzw. Beckengürtel) be-
trifft. Manchmal besteht auch eine andauernde Mitochondriale Enzephalomyopathie mit
Lähmung dieser Muskeln. Pigmentstörungen an Laktatazidose und schlaganfallähnlichen
der Netzhaut im Auge (Retinopathie) können zu Episoden (MELAS-Syndrom)
einer verminderten Sehschärfe, Gesichtsfeldein- Bei dieser Mitochondriopathie ist vor allem das
schränkungen und Blendempfindlichkeit führen. Gehirn betroffen und es kommt zu schlaganfall-
Sind die Nerven von Armen und Beinen betrof- ähnlichen Beschwerden, die bereits bei jungen
fen, so kommt es besonders zu Taubheitsgefüh- Menschen auftreten. Es können plötzliche
len in Füßen und Händen sowie zu Schwindel Lähmungen einer Körperseite, aber auch Sehstö-
(sensible Ataxie). Weitere mögliche Störungen rungen auf einer Seite des Gesichtsfeldes (nicht
sind: Zuckerkrankheit (Diabetes mellitus), Reiz- eines Auges) auftreten. Diese verschwinden
leitungsstörungen im Herz, Kleinhirnschädigung demnach nicht, wenn man ein Auge zukneift.
mit Gleichgewichtsstörungen und Schwindel, Im Unterschied zum klassischen Schlaganfall,
Schwerhörigkeit. Tritt die Erkrankung bereits in der typischerweise schmerzlos ist, haben die
7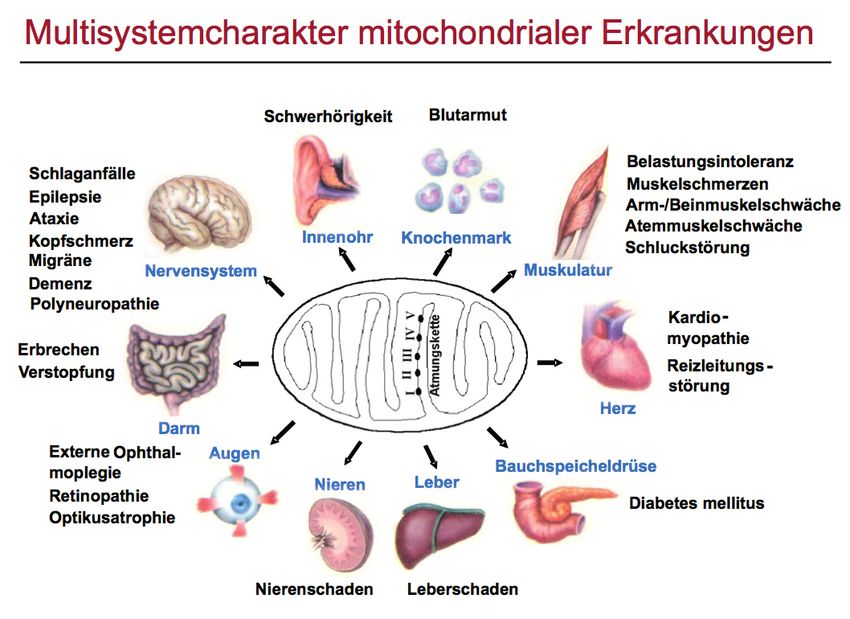
Patienten dabei häufig Kopfschmerzen und auch Myoklonusepilepsie mit Ragged-
Übelkeit und Erbrechen. Außerdem kommt es red-Fasern (MERRF-Syndrom)
dabei vielfach auch zu epileptischen Anfällen. Bei dieser recht seltenen Mitochondriopathie
Charakteristische Laborverändung ist eine sog. stehen epileptische Anfälle und kurze Muskelzu-
Laktatazidose (siehe Diagnose). ckungen (Myoklonien) im Vordergrund. Zusätz-
Auch bei dieser Erkrankung finden sich häufig lich namensgebend waren hier Veränderungen
zusätzlich ganz andere Beschwerden. Typisch in der Muskelbiopsie, die sog. Ragged-red-Fasern
sind Schwerhörigkeit, Diabetes, Kleinwuchs, (siehe unter Diagnostik). Diese Veränderungen
Belastungsintoleranz und Muskelschwäche sowie sind aber nicht typisch für das MERRF-Syndrom
Abbau der geistigen Leistungsfähigkeit (Demenz). und finden sich auch bei vielen anderen Mito-
Auch eine hypertrophe Kardiomyopathie kann chondriopathien, z. B. auch bei der CPEO und
vorliegen. Diese Beschwerden finden sich aber beim MELAS-Syndrom. Ähnlich wie bei diesen
nur bei einem Teil der Patienten. Die Erkrankung beiden Erkrankungen finden sich beim MERRF-
kann aufgrund der schlaganfallähnlichen Episo- Syndrom zusätzliche multisystemische Beschwer-
den in krisenhaften „Schüben“ verlaufen, von den. Die Vererbung ist ähnlich wie beim MELAS-
denen sich die Patienten nicht immer wieder Syndrom, also von der Mutter auf Kinder, bedingt
vollständig erholen. Andere multisystemische durch eine Punktmutation der mitochondrialen
Beschwerden wie z. B. die Schwerhörigkeit neh- Erbsubstanz (meist Mutation m.8344A>G).
men typischerweise im Verlauf der Erkrankung
langsam kontinuierlich zu.
Isolierte mitochondriale Myopathie
Das MELAS-Syndrom ist nicht ganz so häufig wie Es gibt auch Mitochondriopathien, die sich durch
die CPEO. Die Erkrankung beruht auf einer sog. eine Muskelerkrankung (Myopathie) zeigen,
Punktmutation der mitochondrialen Erbsub- die nur die Muskeln von Armen, Beinen oder
stanz (meist handelt es sich um die Mutation Rumpf betrifft und weder eine Schwäche der
m.3243A>G), die von der Mutter an Kinder ver- Augenmuskeln noch Störungen anderer Organe
erbt wird. Vielfach tragen die Mütter von Kindern aufweisen. Manchmal handelt es sich um eine
mit MELAS zwar die Mutation in sich, weisen belastungsabhängige Symptomatik, die zu keiner
aber nur geringe Beschwerden (z. B. eine leichte bleibenden Lähmung führt und als psychisch
Schwerhörigkeit oder eine Zuckerkrankheit) auf. bedingter Erschöpfungszustand verkannt werden
Es ist wichtig zu betonen, dass das bloße Vorlie- kann. Bei diesen isolierten mitochondrialen Myo-
gen dieser Mutation m.3243A>G nicht bedeutet, pathien sind die Gendefekte und die Vererbung
dass das Vollbild eines MELAS-Syndroms mit sehr variabel.
Schlaganfall sich entwickeln wird. Nur bei einem
Teil der Patienten kommt es dazu. Vielfach liegt
in den Familien diese Mutation in unterschiedli- Lebersche Optikusneuropathie
chem Ausmaß (Heteroplasmiegrad) vor und Fa- Auf der anderen Seite gibt es eine Mitochondrio-
milienmitglieder sind unterschiedlich betroffen, pathie, die sich meist ausschließlich am Seh-
oft nur milde oder gar asymptomatisch. nerv (Nervus opticus) abspielt – die Lebersche
Optikusneuropathie. Diese Erkrankung hat ihren
MRT (Kernspin- Namen vom Heidelberger Augenarzt Theodor
tomographie) als Leber, der sie 1871 erstmals in der medizinischen
Diffusionsrichtung von Fachliteratur beschrieb. Typischerweise kommt
einem Patienten mit es bei jungen Männern innerhalb von wenigen
MELAS-Syndrom:
Wochen zu einer schmerzlosen Erblindung (oder
In der rechten Gehirn-
hälfte (linke Bildseite) hochgradigen Minderung des Sehvermögens)
erkennt man eine auf einem Auge, wenige Wochen später folgt das
hellere Darstellung der andere Auge. Bei wenigen Patienten kommt es in
Hirnrinde. den ersten Jahren nach Erkrankungsbeginn auch
8
wieder zu einer spontanen Besserung des Seh- tanz bedingt. Diese werden zwar von der Mutter
vermögens. Viele Patienten bleiben aber leider auf die Kinder vererbt, die Mütter selbst sind
praktisch blind. aber meist beschwerdefrei, da Männer etwa
Die Lebersche Optikusneuropathie wird durch 5-mal häufiger erkranken als Frauen.
Punktmutationen der mitochondrialen Erbsubs-
Mitochondriale Erkrankungen rungen, die durch Schädigung der Netzhaut oder
im Kindesalter des Sehnerven entstehen können. Am Muskel
kann eine Reduzierung der Muskelkraft, der
Prof. Dr. med. Peter Freisinger, Reutlingen Muskelspannung (Tonus), aber auch ein Abbau
Prof. Dr. med. Wolfgang Sperl, Salzburg der Muskelzellen (Rhabdomyolyse) beobachtet
werden.
Mitochondriale Erkrankungen treten im Kindes-
alter zu unterschiedlichen Zeitpunkten auf. So Typische Symptome an anderen Organen sind
gibt es Erkrankungen, die schon bei der Geburt die Kardiomyopathie, d. h. eine Schwäche des
zu erkennen sind, häufiger beginnen die ersten Herzmuskels oder Herzrhythmusstörungen, Le-
Krankheitssymptome im Säuglingsalter. Aller- berschwäche, bzw. -versagen, Niereninsuffizienz
dings gibt es auch Formen, die sich erst im Kin- und andere. Auch die Bildung der Blutzellen kann
des- und Jugendalter zeigen. Die Regel „je früher beeinträchtigt sein (Knochenmarkinsuffizienz).
der Beginn, desto schwerer der Verlauf“ trifft
sehr häufig, jedoch nicht ausnahmslos zu. Im Blut und auch im Gehirn findet sich meist
eine deutliche Erhöhung der Milchsäure (Lak-
Im Gegensatz zu den Erwachsenen leiden Kinder tat), die zu einer Übersäuerung des Organismus
und Jugendliche selten an den typischen mito- führen kann. Es ist wichtig zu wissen, dass nur
chondrialen Syndromen wie z.B. MELAS, etc. selten alle Symptome bei einem Krankheitsbild
(siehe vorhergehender Abschnitt). Viel häufiger auftreten. Häufig finden sich unterschiedliche
zeigen Kinder eine Kombination unspezifischer Kombinationen von mehreren dieser Krankheits-
Symptome, die mehrere Organe betreffen. Des- zeichen.
halb wird die Diagnose häufig mit Verzögerung
gestellt. Da Mitochondriopathien zu Energie-
mangel führen und jede Zelle mehr oder weniger Ursachen
Energie benötigt, betreffen diese Erkrankungen Die genetischen Ursachen von mitochondrialen
die Organsysteme in unterschiedlichem Ausmaß. Erkrankungen im Kindesalter sind sehr vielfältig
Dementsprechend können die Symptome auch und nur zum Teil bekannt. Im Gegensatz zu den
sehr vielfältig sein. Mitochondriopathien im Erwachsenenalter, die
überwiegend durch Veränderungen in der Erb-
substanz der Mitochondrien selbst bedingt sind
Symptome (mitochondriale DNA), liegen bei den kindlichen
Wir unterscheiden Symptome des Nervensys- Erkrankungen häufiger Mutationen der Erb-
tems, der Muskulatur und Symptome an anderen substanz im Zellkern (nukleäre DNA) vor. In den
Organen. Typische mitochondriale Symptome am letzten Jahren wurden sehr große Fortschritte
zentralen Nervensystem (Gehirn) sind Störun- in der Genetik der kindlichen Mitochondrio-
gen der Bewegungskoordination (Ataxie, Dysto- pathien erzielt. Inzwischen sind mehr als 300
nie), Schluck- und Sprachstörungen, Störungen unterschiedliche Krankheitsgene bekannt, die
der geistigen Entwicklung, epileptische Anfälle eine Mitochondriopathie verursachen können.
Innenohrschwerhörigkeit-/Taubheit, und Sehstö- Bei einem Teil der Krankheiten ist die genetische
9
Ursache immer noch unklar. Durch weitere Fort- Weitere neurologische Zeichen sind mangelnde
schritte bei den modernen genetischen Tech- Muskelspannung, Nystagmus (Augenzittern) und
niken (Next Generation Sequencing) ist jedoch eine generalisierte Störung in der Bewegungs-
damit zu rechnen, dass dieser Anteil weiterhin steuerung (Ataxie).
abnimmt.
Oft bleibt das Wachstum zurück (Kleinwuchs)
Die meisten Mitochondriopathien im Kindesalter und es sind andere Organsysteme wie das Herz,
sind mit einer Störung der Zellatmung (Atmungs- die Niere und die Leber beteiligt. Durch die
kette) vergesellschaftet. Es finden sich entweder Schwäche der Muskulatur, aber auch durch die
einzelne oder kombinierte Defekte der unter- Veränderungen in tiefen Regionen des Gehirns
schiedlichen Atmungskettenkomplexe (Komplex (Atemzentrum), kann es zu teils schwerwiegen-
I-V) oder auch im Pyruvatdehydrogenasekomplex den Schluck- und Atmungsstörungen kommen.
(PDHC). Dies führt zum einen zu einem Mangel Die meisten Patienten haben auch eine Übersäu-
an ATP, dem wichtigsten „Brennstoff“ der Zelle, erung mit Laktat.
zum anderen zu einer Anhäufung von „freien
Radikalen“ (toxischen Sauerstoffradikale), die In der Kernspinuntersuchung (MRT) des Gehirns
die Zellwand und Zellbestandteile schädigen und zeigen die Patienten typische, symmetrische Ver-
eine beschleunigte Zellalterung unterstützen. änderungen im Mittelhirn (Basalganglien) und im
Hirnstamm. Diese Veränderungen sind bedingt
Die Atmungskettenkomplexe lassen sich am durch einen Untergang von besonders energie-
besten im Muskelgewebe nachweisen, weshalb abhängigem Gewebe in diesen Bereichen.
früher häufig eine Muskelbiopsie notwendig war.
Durch die Fortschritte in der genetischen Diag- Das Leigh-Syndrom hat sehr viele unterschied-
nostik ist eine Muskelbiopsie nur noch in ausge- liche Ursachen. Inzwischen sind Defekte in
wählten Fällen notwendig. mindestens 80 unterschiedlichen Genen bekannt
In der Muskelbiopsie können sich Veränderungen
Im Folgenden werden die wenigen typischen in allen Atmungskettenkomplexen (I-V) sowie im
mitochondrialen Syndrome im Kindesalter kurz Pyruvatdehydrogenasekomplex (PDHC) finden.
dargestellt. Wie bei der überwiegenden Mehrzahl der mito-
chondrialen Erkrankungen, gibt es bisher für das
Leigh-Syndrom oder Leigh-Erkrankung Leigh Syndrom nur in Einzelfällen eine spezifische
Das Leigh-Syndrom ist keine eigene Erkrankung, Therapie. Die Behandlung umfasst in erster Linie
sondern eine Kombination von typischen Krank- die Therapie der verschiedenen Komplikationen
heitszeichen mit einem typischen Verlauf. Es ist (z. B. Epilepsie, Herzinsuffizienz, Ateminsuffizienz,
eine fortschreitende, überwiegend neurologi- etc. s. Tabelle 1) sowie die Gabe von Vitaminen
sche Erkrankung, die meist im Säuglingsalter, und Cofaktoren (z.B. Thiamin, Coenzym Q10,
manchmal auch etwas später, beginnt. Die ersten Riboflavin u.a.), die je nachdem, welcher
Symptome treten meist nach Infekten auf, wobei Atmungskettenkomplex betroffen ist, gegeben
diese nicht die Ursache sind. Typischerweise werden und die z.T. zu einer Verbesserung der
verlieren die Patienten bereits erlernte Fähigkei- verbleibenden Aktivität dieser Enzyme führen
ten wie z. B. das Halten des Kopfes, Gehen oder kann. Bei Patienten mit Leigh Syndrom und
Sprechen. Dies ist häufig begleitet von anderen einem PDHC Defekt (E1) gibt es in einigen Fällen
Zeichen wie Appetitverlust, häufigem Erbrechen ein Ansprechen mit klinischer Verbesserung auf
und Krampfanfälle. Die Erkrankung verläuft oft Thiamin, aber vor allem auch auf eine ketogene
krisenhaft, d.h. es kommt nach Infekten oder Diät. Leider ist eine Heilung auch damit nicht
Belastungssituationen zu einer akuten Ver- möglich, die Prognose für Patienten mit Leigh
schlechterung, von der sich die Kinder wieder Syndrom ist in den meisten Fällen sehr ernst.
erholen können. Meist erreichen sie aber nicht
mehr das gleiche Niveau wie vor der Krise.
10Diese Erkrankungen können unterschiedliche
Organsysteme betreffen. Häufig ist die Kombi-
nation einer Störung des Gehirns und der Leber
(„hepatocerebral“ s.u.) und des Gehirns und der
Muskulatur („muskulocerebral“). Die häufigste
Krankheitsform ist das Alpers-Syndrom (Synonym:
progressive infantile Poliodystrophie). Diese Er-
A B
krankung ist gekennzeichnet durch eine frühzeitig
auftretende meist therapieresistente Epilepsie,
Kernspintomographie (MRT) des Gehirnes eines Patien- eine ausgeprägte mentale (geistige) Entwicklungs-
ten mit Leigh-Syndrom (A) im Vergleich zu einem störung, Spastik sowie eine zunehmende Stö-
Gesunden (B). Deutlich zu erkennen sind die symet-
rung der Leberfunktion, die sehr unterschiedlich
rischen Aufhellungen im Bereich der Basalganglien
(Pfeil). Diese Aufhellungen sind Zeichen einer Gewebs- ausgeprägt sein kann. Meist haben die Patienten
schädigung infolge des Energiemangels auch eine ausgeprägte Gedeihstörung. Die Ursa-
che für das Alpers-Syndrom sind Mutationen des
Enzyms „Polymerase Gamma 1“ (POLG1). Es gibt
allerdings auch Mutationen in POLG1, die nur zu
Pearson-Syndrom einer Epilepsie führen können, ohne dass andere
Beim Pearson-Syndrom fallen die Patienten im Organsysteme beteiligt sind bzw. andere Fälle, in
Säuglingsalter durch Blutarmut auf, die sowohl denen nur die Leber betroffen ist. Bei einem wei-
die roten als auch die weißen Blutzellen betref- teren hepatocerebralen Syndrom ist der Mangel
fen kann. Zudem haben sie häufig eine einge- der mitochondrialen DNA durch Mutationen in
schränkte Funktion der Bauchspeicheldrüse dem Enzym DGUOK bedingt. Die Patienten fallen
sowie eine ausgeprägte Beeinträchtigung des typischerweise Tage bis Wochen nach der Ge-
Gedeihens. Die geistige Entwicklung ist meist burt durch ein beginnendes Leberversagen und
verzögert. Der Verlauf der Erkrankung ist unter- Störungen der Hirnfunktion auf. Der Verlauf wird
schiedlich stark ausgeprägt. Wird das Erwach- durch das zunehmende Leberversagen bestimmt.
senenalter erreicht, kann das Pearson-Syndrom Bei einer kleinen Gruppe von Patienten mit weit-
häufig in ein Kearns-Sayre-Syndrom (KSS, s. dort) gehend normaler Gehirnfunktion kann eine Leber-
übergehen. Beim Pearson-Syndrom ist die Ursa- transplantation als Therapie erwogen werden.
che eine Deletion in der mitochondrialen DNA,
d.h. ein größeres Bruchstück der mitochondria-
len Erbsubstanz ist verloren gegangen. Erkrankungen durch Störung des
Fettsäureabbaus und andere Störungen
im mitochondrialen Stoffwechsel
Erkrankungen mit Mangel an mitochondrialer
Erbsubstanz (mitochondriale DNA-Depletions- Mitochondrien enthalten neben der lebenswich-
Syndrome) tigen sauerstoffabhängigen Energiegewinnung
(oxidativen Phosphorylierung = OXPHOS) noch
Bei den mitochondrialen DNA-Depletions-Syn- zahlreiche andere wichtige Stoffwechselvorgänge
dromen entsteht durch einen erblichen Defekt wie die β-Oxidation von Fettsäuren, Hämbio-
ein Mangel an mitochondrialer DNA, der wie- synthese, Harnstoffzyklus, Aminosäuresynthese,
derum zu Störungen in der Atmungskette führt. Purin-, Pyrimidinbiosynthese, Cholesterolstoff-
Somit entsteht ein Energiemangel. Die Verer- wechsel, Neurotransmitterstoffwechsel etc. In all
bung ist autosomal rezessiv, d.h. in der Regel diesen Stoffwechselbereichen sind Krankheiten
sind die Eltern symptomfreie Träger. Kinder sind beschrieben, die wir aber nicht im engeren Sinne
mit einer Wahrscheinlichkeit von 25% betroffen. zu den Mitochondriopathien zählen. Sie werden
Wir kennen heute mindestens 10 verschiedene daher auch in dieser Informationsbroschüre
genetische Ursachen. nicht miterfasst.
11Diagnostik
PD Dr. med. Dr. Saskia B. Wortmann, Es gibt kein standardisiertes Vorgehen bei dem
München und Salzburg Verdacht auf eine Mitochondriopathie, denn die
Dr. rer. nat. Holger Prokisch, München Symptome sind vielfältig und die diagnostischen
Möglichkeiten verändern sich rasant. Generell
sollte eine gründliche und ausführliche Anam-
Mitochondrien sind in jeder Zelle unseres nese, inklusive Erhebung des Stammbaums über
Körpers vorhanden, sie werden auch als „Zell- drei Generationen, erfolgen. Hierbei sollte gezielt
kraftwerk“ bezeichnet und produzieren unsere nach häufigen, aber oft übersehenen Sympto-
Energie (ATP). Als mitochondriale Erkrankungen, men einer Mitochondriopathie gefragt werden
auch Mitochondriopathien genannt, werden (siehe Kapitel Krankheitsbilder/Symptome).
genetisch bedingte Erkrankungen der Energie- Bei der körperlichen und neurologischen Unter-
Produktion bezeichnet. Diese Energieproduktion suchung sollte der Fokus auf den verschiedenen
besteht aus diversen Zwischenschritten (z. B. Organen liegen (siehe Kapitel Krankheitsbilder/
Zitratzyklus, Atmungskette), an dem mehr als Symptome). Die Untersuchung der Organbetei-
1.500 Enzyme direkt oder indirekt beteiligt sind. ligung spiegelt sich auch in der Labordiagnostik
Eine Besonderheit dieser Mitochondrienproteine wieder (Blutbild, Transaminasen, Harnstoff, Kre-
ist, dass ihre genetische Information teils auf der atinin, wenn möglich ergänzt durch ein Amino-
DNA des Zellkerns, teils auf der mitochondrialen säurenprofil in Blut und Urin sowie organischen
DNA gespeichert ist. Momentan sind genetische Säuren und Purine/Pyrimidine im Urin). Häufig
Fehler (Mutationen) in mehr als 290 Genen ist bei einer Mitochondriopathie der Milchsäure-
bekannt, die zu einer Mitochondriopathie führen wert (Laktat) im Blut und/oder im Gehirnwasser
können. (Liquor) erhöht. Dieser sollte daher mehrmals
gemessen werden. Es ist darauf zu achten, dass
Da die Mitochondrien in jeder Körperzelle vor- die Blutabnahme ungestaut erfolgt, da das
handen sind, kann sich eine Mitochondriopathie Stauen zu einem fälschlich erhöhten Wert führen
mit Symptomen an jedem Organ präsentieren, kann. Manchmal ist es auch sinnvoll, das Laktat
in unterschiedlicher Kombination und in jedem nach kontrollierter körperlicher Anstrengung
Alter. Organe mit besonders hohem Energiever- (z. B. Fahrrad-Ergometrie) zu messen.
brauch (Skelettmuskel, Herzmuskel, Gehirn) sind
besonders anfällig für Störungen des Energie- Je nach (vermuteter) Organbeteiligung sollten
haushalts und zeigen demzufolge auch häufiger Untersuchungen durch den Augenarzt (Untersu-
Symptome (z. B. Muskelschmerzen, Herzmus- chung des Augenhintergrunds), ein Hörtest und
kelerkrankungen, Epilepsie etc.). Diese Vielfalt eine kardiologische Untersuchung (EKG, Langzeit-
an Symptomen und Symptomkombinationen EKG, Herzultraschall) erfolgen. Da bei Mitochon-
erschwert häufig die schnelle Diagnose einer driopathien sehr oft das zentrale oder periphere
Mitochondriopathie. Um weitere Verzögerun- Nervensystem betroffen ist, erfolgen auf diesem
gen bei der Diagnosestellung und überflüssige Gebiet häufig besonders viele zusätzliche Unter-
Untersuchungen zu vermeiden, sollte die weitere suchungen (Magnetresonanztomographie (MRT)
Diagnostik in Zusammenarbeit mit einem spe- des Gehirns, Messung der Gehirnströme (EEG),
zialisierten Zentrum erfolgen. Adressen dieser Messung der Nervenleitgeschwindigkeit (Elektro-
Neuromuskulären Zentren mit dem Schwerpunkt neurographie) etc.
mitochondriale Erkrankungen können bei der Während früher im Anschluss in der Regel eine
Deutschen Gesellschaft für Muskelkranke e. V. Muskelbiospie erfolgte („biopsy first“), wird ein
(DGM), Webseite: www.dgm.org, erfragt wer- spezialisiertes Zentrum heute eine genetische
den. Jede einzelne Mitochondriopathie (jeder Untersuchung durchführen („genetics first“).
einzelne genetische Defekt) ist selten, in ihrer Dieses Vorgehen hat viele Vorteile: Zunächst
Gesamtheit allerdings weniger, denn einer von entfallen die Schmerzen nach einer Muskelbi-
ca. 4.500 Menschen leidet an einer mitochondri- opsie und bei Kindern auch die hierfür nötige
alen Erkrankung. Narkose. Außerdem ist die Aussagekraft einer
12Muskelbiopsie begrenzt, besonders bei Kin- rung, nahezu alle wichtigen Genabschnitte des
dern. Bei Erwachsenen können in vielen Fällen Menschen in einem Test mit nur ca. 5 ml Blut zu
lichtmikroskopisch charakteristische Zeichen untersuchen.
einer Mitochondriopathie erkannt werden (z. B.
ragged-red-fibres – RRF), bei Kindern findet man Einschränkend ist zu erwähnen, dass sich trotz
diese jedoch sehr selten. Es ist des Weiteren der stark verbesserten Technik nur ca. 50% aller
möglich, durch die Muskelbiopsie die Aktivität Mitochondriopathien genetisch sichern lassen.
der verschiedenen Proteine der Atmungskette zu Gelingt es nicht, mit dem genetischen Test eine
messen und so einzugrenzen, welcher Schritt der Diagnose zu stellen, kann trotzdem noch eine
Energieproduktion gestört ist. Damit kann die Muskelbiopsie sinnvoll sein, um die Erkrankung
Muskelbiopsie heute die genetische Diagnostik zumindest auf biochemischem Niveau zu diag-
unterstützen, aber nicht ersetzen. nostizieren.
Während früher in der genetischen Diagnostik Generell sollte parallel zur Blutprobe für die
nur in jeweils einem Gen nach genetischen Feh- genetische Untersuchung auch eine Hautbiopsie
lern gesucht werden konnte – und damit die Zahl zur Gewinnung von Fibroblasten (Zellen im Bin-
der in Frage kommenden Kandidatengene durch degewebe) vorgenommen werden. In diesen Zel-
die vorherige Muskelbiopsie sinnvoll begrenzt len können die Atmungskettenproteine ebenfalls
wurde – ermöglicht die genetische Untersuchung biochemisch untersucht werden. Die Hautbiopsie
der neuesten Generation, die Exomsequenzie- kann auch bei Kindern ohne Narkose durchge-
13führt werden. Desweiteren sind zusätzliche Tests, wohl fehlerhafte als auch fehlerfreie Kopien.
z. B. wenn eine bisher unbekannte genetische Dieses Phänomen wird als Heteroplasmie
Veränderung gefunden wird, in Fibroblasten gut bezeichnet, tragen alle Kopien den Fehler spricht
möglich. man von Homoplasmie. Im Allgemeinen ist die
Anzahl der fehlerhaften Kopien in dem Körper-
Es sollte noch erwähnt werden, dass die Diagnos- gewebe/Organ am höchsten, welches auch am
tik von Veränderungen in der mitochondrialen stärksten betroffen ist. Im Fall einer Muskeler-
DNA eine Besonderheit darstellt. Im Gegensatz krankung aufgrund eines Fehlers in der mito-
zu der DNA, die im Zellkern als Chromosomen chondrialen DNA, kann eine Muskelbiopsie erfor-
vorliegt, ist die mitochondriale DNA klein und derlich sein, um die genetische Untersuchung im
ringförmig angelegt. In jeder Zelle gibt es viele am meisten betroffenen Gewebe durchzuführen.
Kopien dieser mitochondrialen DNA. Wenn eine
Mitochondriopathie vorliegt, gibt es meist so-
14Übersicht zu den Therapiemöglichkeiten
Prof. Dr. Marcus Deschauer, München (z. B. Coenzym Q, Thiamin = Vitamin B1, Ribo-
Prof. Dr. Peter Freisinger, Reutlingen flavin = Vitamin B2, Biotin und Carnitin) klar
Prof. Dr. Wolfgang Sperl, Salzburg indiziert ist. Diese Defekte führen meist zu einer
Mitochondriopathie mit Beginn im Kindesalter.
Eine Heilung mitochondrialer Erkrankungen Bei Störungen des Fettsäureabbaus sollte eine
ist in der Regel nicht möglich und auch derzeit kohlenhydratreiche Ernährung bevorzugt wer-
noch nicht absehbar. Selbst wenn der Gende- den, bzw. je nach Enzymdefekt eine Diät mit
fekt bekannt ist, kann er nicht behoben werden. mittelkettigen Fettsäuren eingehalten werden.
Forschungen mit dem Fernziel einer Gentherapie Starke körperliche Aktivitäten und Fasten sollten
werden derzeit unternommen. vermieden werden. Sollte es zu einer Attacke mit
Muskelzerfall (Rhabdomyolyse) kommen, die sich
Medikamentös kann man versuchen, durch ein durch dunkelbraunen Urin und heftige Muskel-
Mehrangebot bestimmter Substanzen, die für die schmerzen zeigen kann, muss sofort ein Arzt
Zellatmung erforderlich sind, das Energieangebot konsultiert werden. Es muss reichlich Flüssigkeit
der Zellen zu verbessern. Zusätzlich wirken einige zugeführt werden, um die Niere zu spülen, in der
dieser Substanzen (z. B. Coenzym Q10) als sog. sich das Abbauprodukt „Myoglobin“ ablagern
Antioxidantien und können möglicherweise eine kann. In besonders schweren Fällen kann auch
Schädigung der Mitochondrien durch aggressive eine Dialyse (Blutwäsche) erforderlich sein. Bei
Sauerstoff-Formen vermindern. Verschiedene Störungen der Zellatmung kann leichtes körper-
Substanzen wurden in unterschiedlichen Kom- liches Ausdauertraining die Belastungsschwäche
binationen eingesetzt: Coenzym Q, Creatin, verbessern.
Carnitin, Vitamin B1, B2, C, E und K. Eindeutige
Wirkungen konnten bisher nur bei einzelnen Bei den Mitochondriopathien in allen Altersstu-
Mitochondriopathien bewiesen werden. Da nen- fen kommen unterstützende Maßnahmen eine
nenswerte Nebenwirkungen durch Vitamine und große Bedeutung zu. Bei Patienten mit Herz-
Co-Faktoren nicht zu befürchten sind, ist ein The- rhythmusstörungen ist die rechtzeitige Anlage
rapieversuch über wenige Monate gerechtfertigt, eines Herzschrittmachers wichtig (s. Kapitel 10).
auch wenn eine Wirksamkeit nicht bewiesen ist. Ein Hängen der Lider (Ptose) kann vom Augen-
In Einzelfällen profitieren Patienten sehr von den arzt operativ korrigiert werden, wenn die Mus-
beschriebenen Substanzen, bzw. es gibt deutli- keln, die für den Augenschluss verantwortlich
che Hinweise auf deren Wirksamkeit. sind, kräftig sind (s. Kapitel 8). Hörstörungen
können durch ein Hörgerät gebessert werden,
Idebenone (Raxone®) ist zur Behandlung der in Einzelfällen kann auch ein Cochlea-Implantat
Leberschen Optikus-Neuropathie zugelassen und indiziert sein. Bei schwerer Myopathie kann
führt zu einer besseren Erholung der Sehschärfe. einer nächtlichen Hypoventilation (Minder-
Patienten, meistens Kinder, mit einem PDHC- atmung) durch eine nicht-invasive häusliche
Mangel profitieren von einer ketogenen Diät, da Maskenbeatmung entgegengewirkt werden (s.
der defekte Pyruvatdehydrogenasekomplex um- Kapitel Atmungsprobleme). Schluckstörungen
gangen werden kann. Manche der PDHC-Defekte können logopädisch behandelt werden (s. Kapitel
sind auch noch durch Vitamin B1 (Thiamin) be- 9). Patienten mit Mitochondrien-Erkrankungen
einflussbar. Es gibt auch sehr seltene Mitochon- können auf bestimmte Medikamente empfindli-
driopathien (ECHS1-Mangel, HIBCH-Mangel), die cher reagieren, insbesondere auf Narkosemittel
auf eine spezielle Diät mit verminderter Zufuhr (s. Kapitel 12). Bei der medikamentösen Behand-
der Aminosäure Valin ansprechen können. In lung epileptischer Anfälle sollte auf Valproat und
den vergangenen Jahren wurden außerdem eine Barbiturate verzichtet werden, da dieses die
ganze Reihe sehr seltener kofaktorabhängiger Mitochondrienfunktion beeinträchtigen können.
genetischer Defekte beschrieben, bei denen eine Die Behandlung eines Diabetes mellitus erfolgt
gezielte Substitution des betroffenen Kofaktors nach allgemeinen Richtlinien, auf Metformin
15sollte aber verzichtet werden. Krankengymnastik Tiermodellen in Erprobung und zeigen z. T. gute
und physikalische Therapie sind wichtig für die Ergebnisse. Allerdings wird erst die noch ausste-
Erhaltung der Beweglichkeit und zur Schmerzlin- hende klinische Erprobung zeigen, inwieweit sich
derung (s. Kapitel 6 und 11). die Ergebnisse auf den Menschen übertragen
Einige Substanzen, die die Aktivität der Atmungs- lassen.
kette in den Mitochondrien erhöhen oder zu Ähnliches gilt für gentherapeutische Ansätze für
einer Vermehrung der Anzahl der Mitochondrien einzelne spezifische Defekte.
in den Zellen führen, sind in Zellkulturen oder in
16Mitochondriale Erkrankungen und Lebensqualität –
Langfristige Krankheitsfolgen frühzeitig beachten und behandeln!
Carsten Gamroth; Dipl.-Psych.; und durch Mitarbeiterinnen und Mitarbeiter der
Psychologischer Psychotherapeut; Lübeck Zentralinstituts für Seelische Gesundheit (ZI) in
Mannheim durchgeführt. Zur Drucklegung dieses
Die Patientenselbsthilfe für Mitochondrial Er- Handbuches ist die Erhebung und Auswertung
krankte in Deutschland orientiert sich stark an schon recht weit gediehen und die subjektiven
dem Modell der Weltgesundheitsorganisation Eindrücke von Patienten, auf die wir uns im Dialog
(WHO) „Internationale Klassifikation der Funkti- mit Forschern und Klinikern bislang berufen muss-
onsfähigkeit, Behinderung und Gesundheit“, das ten, sind nun durch harte Fakten belegbar. In na-
Krankheiten als ein Zusammenspiel von körper- her Zukunft werden die Ergebnisse veröffentlicht.
lichen, psychischen und sozialen Faktoren sieht. Die Studie trägt den Namen: „Lebensqualität,
Sie alle zusammen sind bei der Entstehung und Schmerz und neuropsychologische Einschränkun-
für den Verlauf einer Erkrankung bedeutsam. gen bei Patienten mit Mitochondrialen Erkrankun-
Neben den primären biologischen Ursachen und gen“ (Dr. Susanne Becker und Prof. Dr. Herta Flor
Folgen einer Mitochondrialen Erkrankung, bietet in Kooperation mit Dipl.-Psych. Carsten Gamroth)
es damit auch weitere, individuell beeinflussbare
Faktoren an. Diese multifaktorielle Herangehens- Ein Novum ist dabei auch die Kooperation mit
weise bietet den Betroffenen letztendlich viele Wissenschaftlern, die ihren Schwerpunkt auf
Möglichkeiten, auf den Verlauf der Erkrankung sozialmedizinische Fragestellungen ausrichten
und die Schwere einzelner Symptome Einfluss zu und nicht aus dem Konsortium der Experten mit
nehmen – dies ist der Kern von Selbsthilfe- und neurogenetischem Schwerpunkt stammen. Diese
Bewältigungsaktivitäten! Erweiterung des Blickwinkels war von den Betrof-
fenen immer wieder gefordert und lange vermisst
worden.
Warum ist dieser Aspekt so wichtig für
Mito-Betroffene?
In der Erforschung von Mitochondrialen Erkran-
kungen wurde traditionell auf Symptomkomplexe
geschaut, die primär und hauptursächlich anhand
von spezifischen Defekten in der Atmungskette
oder anderen bekannten Gendefekten erklärbar
waren (z. B. Augenmuskelbeteiligung, Herzerkran-
Abb.1 ICF-Modell: kungen, Innenohrschwerhörigkeit, Schlaganfälle
ICF (International Classification of Functioning, usw.). Selbst wenn die Ergebnisse der neuroge-
Disability and Health) Die „Internationale Klassi- netischen Studien zeigten, dass Mitochondriale
fikation der Funktionsfähigkeit, Behinderung und
Erkrankungen i.d.R. einen Multisystemcharakter
Gesundheit.“
aufweisen, so bleibt die grundsätzliche Heran-
Die Diagnosegruppe Mitochondriale Erkrankun- gehensweise aber dennoch unifaktoriell. Das
gen in der DGM (DG) verfolgt seit vielen Jahren bedeutet, man sucht nach bereits bekannten
durch zahlreiche Veranstaltungen und Publikati- Ursachen für Beschwerden, die vom Patienten
onen das Ziel, aktive Bewältigungskompetenzen geklagt werden. Findet man die vermuteten
zu verbessern und zu stabilisieren. Seit 2015 wird Defekte nicht, werden die Beschwerden nicht mit
durch die DGM eine Studie gefördert, die erstma- der nötigen Intensität weiter beachtet und für die
lig systematisch erhebt, welche Beschwerden und Betroffenen entsteht oft das Gefühl, man glaube
Symptome bei den Betroffenen zu den größten ihnen nicht. Multifaktorielle Modelle hingegen
Einschränkungen im persönlichen Alltag führen. sind alles andere als limitierend. Sie sind offen
Diese Studie wurde unter Federführung der DG für die Angaben der Betroffenen und suchen
und der internationalen Patientenorganisation über fachliche Grenzen hinaus nach Lösungs- und
IMP (International Mito Patients) vorangetrieben Therapieansätzen.
17Beispiel 1: zu sehen? Lange schon bekannt und gesichert
Chronische Schmerzen bei sind Symptome wie Belastungsintoleranz, frühe
Mitochondrialen Erkrankungen Erschöpfung, Muskelschwäche etc.! Wenn man
nun bedenkt, dass Mitopatienten eher zu den
Bereits vor 10 Jahren problematisierten Mitobe- ‚aktiven Bewältigern‘ einer schweren chronisch-
troffene auf einem bundesweiten DGM-Fachtag progredienten Erkrankung gehören, dann ist
ihre häufigen und intensiven Schmerzbeschwer- auch nachvollziehbar, dass es unter ständiger
den. Eine spontane Befragung unter den mehr körperlicher und mentaler Überforderung früher
als 200 Teilnehmern zeigte, dass eine überdeutli- oder später auch zu chronischen Schmerzen
che Mehrheit angab, im Alltag wesentlich durch kommt.
die Schmerzbeschwerden eingeschränkt zu sein.
Den Betroffenen und der Selbsthilfe muss es
Nun weisen die Ergebnisse der Mannheimer dabei egal sein, ob eine genetische Grundlage
Wissenschaftler darauf hin, dass Schmerzen der Beschwerden zum jetzigen Zeitpunkt er-
bei über 90% der untersuchten Stichprobe ein kennbar ist oder nicht. Wenn die Betroffenen ein
erhebliches Leiden und Einschränkungen in der Symptom wie Schmerz als wesentlich einschrän-
Lebensführung verursachen. Die Schmerzstärke kend und behindernd angeben, dann entwächst
und Häufigkeit entspricht dabei dem Niveau, daraus die Verpflichtung, dieses Thema intensi-
welches auch von reinen Schmerzpatienten mit ver zu erforschen und nach Hilfsmöglichkeiten zu
unspezifischen Rücken- und Muskelschmerzen suchen!
angegeben wird. Diese Gruppe der Schmerzpa-
tienten in der BRD eignet sich sehr gut, um dar- Was kann man bei chronischen Schmerzen tun?
zustellen, welches enormes Belastungspotential Seit den 70er Jahren gibt es auch in der BRD eine
durch die Schmerzsymptomatik entsteht. spezifische Erforschung und Therapie chroni-
scher Schmerzstörungen. Mittlerweile gibt es für
In Abgrenzung zu den klassischen Schmerzpati- Einrichtungen und Fachtherapeuten (Ärzte und
enten fällt nach ersten Analysen der Studienda- Psychotherapeuten) qualitätsgesicherte Zusatz-
ten aber auf, dass Mitobetroffene ihre Schmerz- ausbildungen und Titel, die es dem Betroffenen
problematik ohne extreme Katastrophisierung ermöglichen, einen Spezialisten für Schmerzer-
und Angst bewältigen. Mitos scheinen den krankungen zu finden. Die Nachfrage nach spe-
Schmerz, wie viele andere Symptome, irgend- zieller Schmerztherapie ist riesig. Daher gilt es
wann zu akzeptieren und versuchen noch sehr auch hier Geduld, Beharrlichkeit und Eigeninitia-
lange aktiv am Alltags- und Berufsleben teilzu- tive einzubringen. Insbesondere in Ballungszen-
nehmen. Trotz einer hohen Belastung schätzen tren gibt es einige Spezialisten und Einrichtun-
Mitopatienten mit chronischen Schmerzen diese gen für die Behandlung von Schmerz. Ärztliche
offenbar nicht als so bedrohlich ein, wie das Schmerztherapeuten sind dabei verpflichtet,
bei anderen Schmerzpatienten ohne seltenen mit anderen Berufsgruppen zu kooperieren, um
Erkrankungshintergrund häufig erlebt wird. Die- der Vielschichtigkeit der Schmerzproblematik
ser Aspekt mag ein Grund dafür sein, dass das gerecht zu werden.
Klagen über Schmerz bei Mitoexperten nur sehr
zögerlich als zentrales Problem wahrgenommen Mitopatienten mit chronischen Schmerzen (=
wurde. Ein anderer Grund liegt sehr wahrschein- dauernder oder wiederkehrender Schmerz in
lich darin, dass die bislang beteiligten Experten einem oder mehreren Organsystemen über den
keinen klassisch-neurologischen Mechanismus Zeitraum von drei Monaten) sollten daher daran
erkennen konnten und daher diese Beschwerden denken, einen Fachmann für die Schmerzbe-
vernachlässigten. handlung aufzusuchen. Da man bei der medika-
mentösen Therapie von Schmerzen bei Patienten
Aber ist es denn so schwer, die Logik hinter den mit einer Mitochondrialen Erkrankung einige
Schmerzfolgen für Mitochondrial Erkrankte Besonderheiten berücksichtigen muss, wäre die
18Vorlage dieses Handbuches (Liste der potenziell Beispiel 2:
gefährlichen Medikamente) zu empfehlen. Auf Stimmungsveränderung und Depressivität
jeden Fall ist der Besuch einer interdisziplinären bei Mitochondrialen Erkrankungen
Schmerzambulanz oder einer Schmerzpraxis, die
auch eine schmerzpsychotherapeutische und Wie bereits ausgeführt, wird im Verlauf einer
-physiotherapeutische Behandlung (= multimo- chronischen Erkrankung immer häufiger auch
dale Therapie) anbietet, zu favorisieren. von psychischen Faktoren gesprochen. Dieses
Thema ist bis heute aber sehr negativ besetzt
Die sicherste Möglichkeit, einen zertifizierten und wird auch von vielen Mitobetroffenen zu-
Schmerztherapeuten zu finden, ergibt sich über nächst abgelehnt.
den Besuch der Internetseiten der Fachgesell- Geht man unvoreingenommen an die Thematik
schaften. Zum Beispiel gibt es unter www.dgss. heran, lässt sich auch hier die Logik weiterer
org (Deutsche Schmerzgesellschaft) eine Such- (sekundärer!?) Krankheitsfolgen schnell erken-
funktion nach Postleitzahl. nen: Mitochondriale Erkrankungen gelten als
Unter www.schmerzpsychotherapie.net findet unheilbar! Mitobetroffene scheinen aber sehr
man auch ständig aktualisierte Listen von Psy- lange ein hohes Niveau an Aktivität und Teilhabe
chotherapeuten, die sich auf die Behandlung von aufrechtzuerhalten.
Schmerzstörungen spezialisiert haben. Diese Ex-
perten wissen, dass die vom Patienten erlebten Trotz möglicherweise guter Bewältigungsressour-
Schmerzen i.d.R. nicht durch psychische Vorgän- cen schreitet die Krankheit aber weiter voran.
ge entstanden sind, im langjährigen Verlauf einer Irgendwann sind dann die Einschränkungen
Erkrankung aber sehr wohl mit Veränderungen unübersehbar. Die Berufsfähigkeit ist in Gefahr,
im Verhalten und Erleben reagiert wird. Dies geliebte Hobbys müssen aufgegeben werden,
wiederum hat einen zumeist negativen Einfluss der Aktivitätsradius wird kleiner, Beziehungen
auf die Intensität und Häufigkeit der Schmerz- verändern sich, Gefühle von Hilflosigkeit und
symptome. Hoffnungslosigkeit melden sich immer häufiger.
All dies sind bekannte Faktoren, die bei allen
Daher empfiehlt die Selbsthilfe allen Mitobe- chronischen Erkrankungen zu Veränderungen in
troffenen, frühzeitig an eine Behandlung durch psycho-sozialen Bereichen führen. Diese Krank-
Spezialtherapeuten für das Leitsymptom (z. B. heitsfolgen haben wiederum Einfluss auf den
Schmerz) zu denken. Einen zertifizierten Spezial- weiteren Verlauf der Erkrankung – man spricht
therapeuten für Mitochondriale Erkrankungen von einem Teufelskreis.
wird es nicht so schnell geben! Daher ist ein ganz
individuelles Puzzle an mehreren Therapiebau- Sehr häufig findet man bei chronisch Kranken
steinen für den Betroffenen anzustreben. Der eine relevante Zunahme an depressiven Sympto-
Hausarzt oder Neurologe sollte in diesem Falle men. Insgesamt schätzt man den Anteil der Men-
eine Lotsenrolle übernehmen und dabei helfen, schen, die in der BRD an Depressionen leiden auf
ein passendes Behandlernetz aufzubauen. 12-15 Prozent. Schaut man sich klinische Stich-
proben von Menschen mit chronischen Erkran-
kungen an, so steigt der Anteil der Depressiven
rasant an. Einige Studien zeigen, dass beispiels-
weise bei Schmerzstörungen im späteren Verlauf
jeder Zweite auch Symptome einer klinisch
relevanten depressiven Störung aufweist.
19Sie können auch lesen

















































