Neuromuskuläre Erkrankungen - Symptome und Syndrome Diagnostische Maßnahmen Klassifikation neuromuskulärer Erkrankungen Ausgewählte ...
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
Neuromuskuläre Erkrankungen
• Symptome und Syndrome
• Diagnostische Maßnahmen
• Klassifikation neuromuskulärer Erkrankungen
• Ausgewählte Erkrankungen und Therapien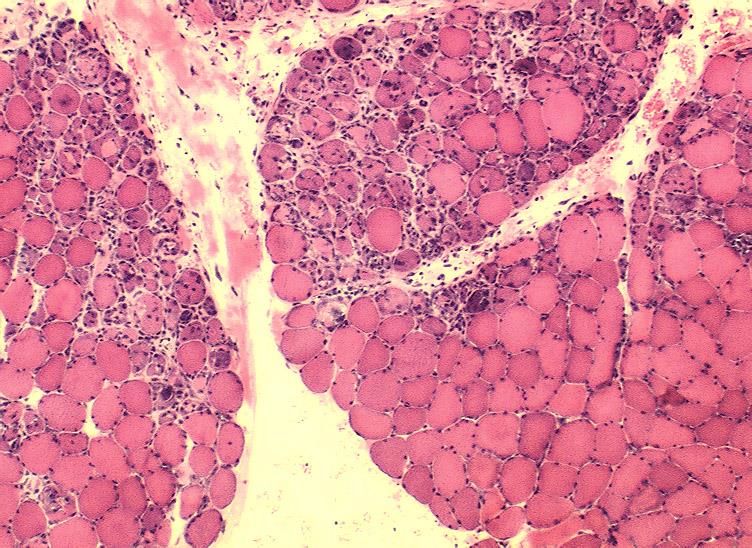
Neuromuskuläre Erkrankungen
-Syndrome-
Myopathisches Syndrom
Paresen, Muskelatrophie oder (Pseudo-)hypertrophie,
Muskeleigenreflexe abgeschwächt (gemäß Muskelmasse), Myalgien,
Muskelsteife, Dekontraktionshemmungsphänomene (Myotonie),
Muskelcrampi, rippling, mounding, oft proximale Betonung,
Muskelenzyme oft erhöht (CK, LDH, GOT, GPT), keine
Sensibilitätsstörungen
Typische Erkrankungen:
Myopathien (entzündlich, degenerativ, hereditär, metabolisch)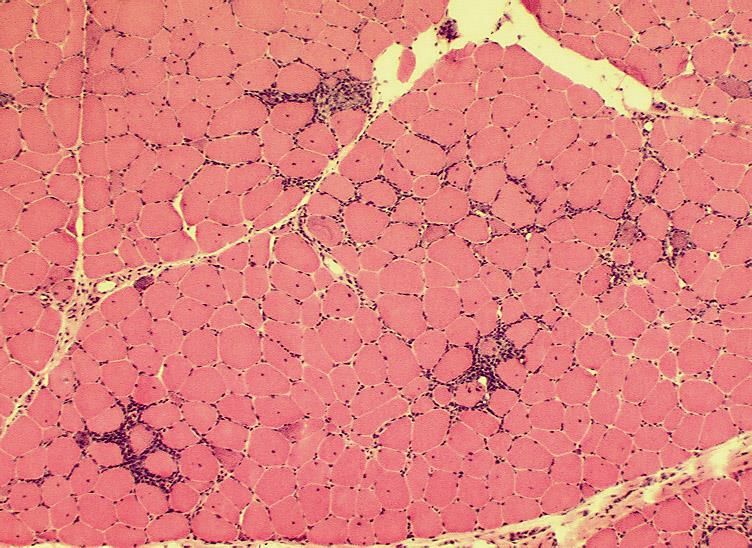
Neuromuskuläre Erkrankungen
-Syndrome-
Neuromuskuläre Übertragungsstörungen
Belastungsabhängige Muskelschwäche, positives
Dekrement, „warming-up“-Phänomen, positives Inkrement,
selten trophischen Störungen des Muskels
Läsionsort:
Motorische Endplattenregion
Typische Erkrankungen:
Myasthenia gravis, Lambert-Eaton-Syndrom (LEMS)
Bei welchen anamnestischen Hinweisen sollte man an eine Myopathie denken? • Vorbekannte „Leberwerterhöhung“ • Auffällige motorische Entwicklung als Kind • Ungewöhnliche Muskelkater- oder Krampfneigung • Muskuläre Belastungsintoleranz • Unklare kardiale Erkrankung/respiratorische Einschränkung • Myoglobinurie-Episoden • Narkosezwischenfälle (Maligne Hyperthermie?) • Familienmitglieder mit CK-Erhöhung, Narkosezwischenfall
Worauf ist bei Erhebung des
neurologischen Status besonders zu achten?
• Distale, proximale oder generalisierte Paresen?
• Muskelatrophien? Hypertrophien?
• Faszikulationen? Myotonie?
• Fazies myopathica? Externe Ophthalmoplegie? Ptosis?
Hoher gotischer Gaumen? Makroglossie?
• Dysphagie? Dysarthrie?
• Dyspnoe? Paradoxe Atmung?
• Skoliose? Kontrakturen?
• Scapula alata?
• Sensibilitätsstörungen?
Klinische „Tests“/ Leitbefunde

Moderne diagnostische Maßnahmen Anamnese, klinische Untersuchung! Labor (ggf. mit Biochemie) CK mit Isotypisierung, Trockenbluttests, Acylcarnitinprofil/Carnitinstatus im Plasma, Antikörperbestimmungen, Schilddrüsenhormone, etc. Elektrophysiologie Muskel-/Nervensonografie Muskel-MRT (Art/Ausmaß der Muskelaffektion) T1 Fett, Trophik; T2, STIR Ödem-ähnliche Veränderungen Diagnostische Zuordnung (Befallsmuster), Richtung der genetischen Aufarbeitung, Auswahl einer geeigneten Biopsiestelle Muskelbiopsie Genetik ? Einzelgen-Sequenzierung nach Sanger → NGS-Techniken mit PANEL-Diagnostik, WES/WGS
Diagnostik bei neuromuskulären Erkrankungen
-Elektrophysiologie-
1. EMG
2. Elektroneurographie
3. Dekrement/Inkrement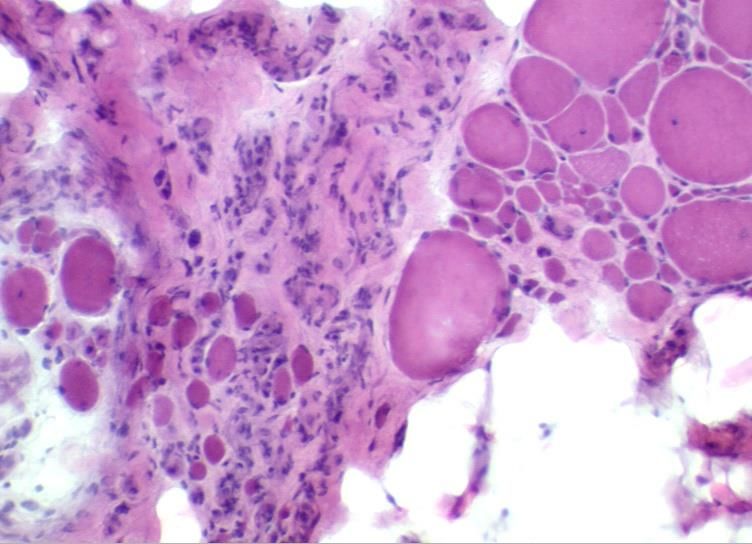
Diagnostik bei neuromuskulären Erkrankungen
-Indikation zur Muskelbiopsie-
V.a. entzündliche Muskelerkrankungen
V.a. bestimmte erbliche Myopathien
V.a. bestimmte metabolische Myopathien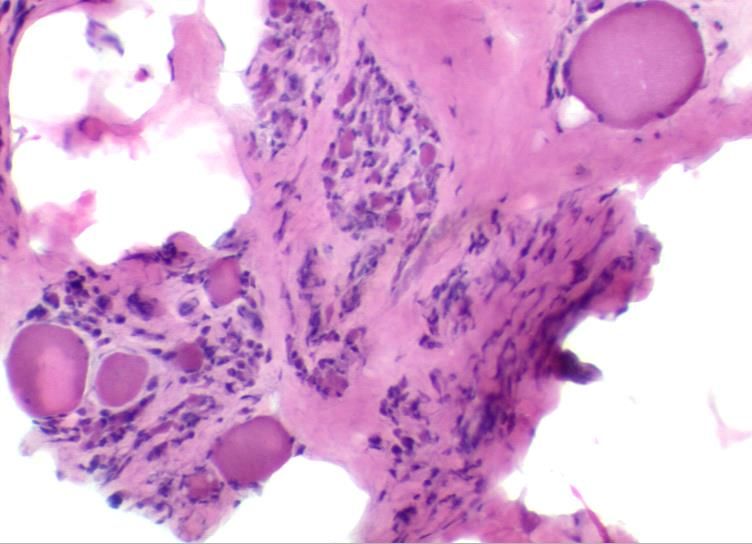
Biopsieverarbeitung - Einfrieren Einfrieren in tauendem Isopentan, dann in flüssigem Stickstoff
Unauffälliger Skelettmuskel H&E
Myositisches Gewebssyndrom H&E
Symptomatische Therapien Kardiologie EKG, 24h-EKG, TTE, Kardio-MRT und ggf. frühzeitig Medikation (z.B. ACE-Hemmer) Pneumologie Lungenfunktion/BGA, kardiorespiratorische Polygrafie, Schlaflabor, non-invasive/invasive Heimbeatmung, Expektorationshilfen, Impfungen Orthopädie (Skoliose, Kontrakturen) Bildgebung („Skoliosimetrie“), Physiotherapie, ggf. OP Ernährungssicherung (Logopädie, ggf. PEG) Hilfstechnische/sozialmedizinische Versorgung Verhaltensempfehlungen Keine extremen Belastungen (eher konzentrisch als exzentrisch), keine myotoxischen Medikamente ohne zwingende Indikation Physio-, Ergotherapie; Logopädie Aufklärung Narkoserisiko (Ausweis)!
Spezifische Therapiekonzepte
Implementierung von Patientenregistern und sog. standards of care
Unmittelbarer Patientennutzen
Schaffung optimaler Voraussetzungen für die Durchführung klinischer Studien
Entwicklung von Orphan drugs für Orphan Diseases
“Orphan drugs expected to be ~20% of worldwide prescription sales by 2020 (excluding generics)”
Diätetische Maßnahmen, Antioxidantien, Enzymersatztherapien, Chaperone
Modulatoren der Autophagie, Proteasom-Regulation, mitochondrialen Biogenese
Gentherapieansätze von herausragender Bedeutung:
Genome Editing mit Reparatur von Gendefekten auf DNA-Ebene (Transcription activator-like effector-Nukleasen
TALEN, sequenzspezifische Restriktionsenzyme; CRISPR/Cas-Systeme)
Antisense Oligonukleotid (AON) Strategien (zielen auf mRNA, liegen sog. Exon Skipping Techniken zugrunde)
in Erforschung weit fortgeschritten, in klinischer Entwicklung
Zahlreiche klinische Studien!Patientenregister
Myotone Dystrophien Typ 1/2 Mitochondriale Erkrankungen
Treat-NMD Patientenregister; Deutschland, Schweiz, Österreich mitoNET mit mitoREGISTER
Dystrophinopathien, SMA, FKRPopathien, CMT Pompe Registry
Treat-NMD Patientenregister
GNE-MyopathienKlassifikation nach klinischen Schwerpunkten
Biopsie (Genetik,
Biochemie?)
„Blickdiagnose“, direkt Genetik! Biopsie Einzelfallentscheidung
DM1? FSHD? OPMD? sIBM? LGMD?
Proximal, distal, axial, fazial, bulbär?
Extramuskuläre Symptome?Andere Klassifikationsmöglichkeiten Nach Muskel-Histologie dystroph myopathisch neurogen entzündlich spezielle Strukturstörung spezielle Speicherung Enzymhistochemie (Aktivitätsnachweise) Immunhistochemie (Proteinnachweise) Nach genetischem Befund
Klassifikation der Myopathien Muskeldystrophien (z.B. Dystrophinopathien Duchenne/Becker, FSHD) Strukturmyopathien (kongenitale/Proteinaggregat-Myopathien) Endokrine/toxische Myopathien Myotone Dystrophien (z.B. Myotone Dystrophie Typ 1) Ionenkanalerkrankungen (Myotonien) Metabolische Myopathien (z.B. M. Pompe) Mitochondriale Erkrankungen Entzündliche/immunvermittelte Myopathien (Myositiden)
Dystrophinopathien
Häufigste Muskeldystrophien
X-chromosomal rezessiv
Phänotypen:
DMD, Muskeldystrophie Typ Becker BMD,
symptomatische Konduktorinnen
Prävalenz DMD 1:3.500 m Neugeborene
Prävalenz BMD 1:30.000 m Neugeborene
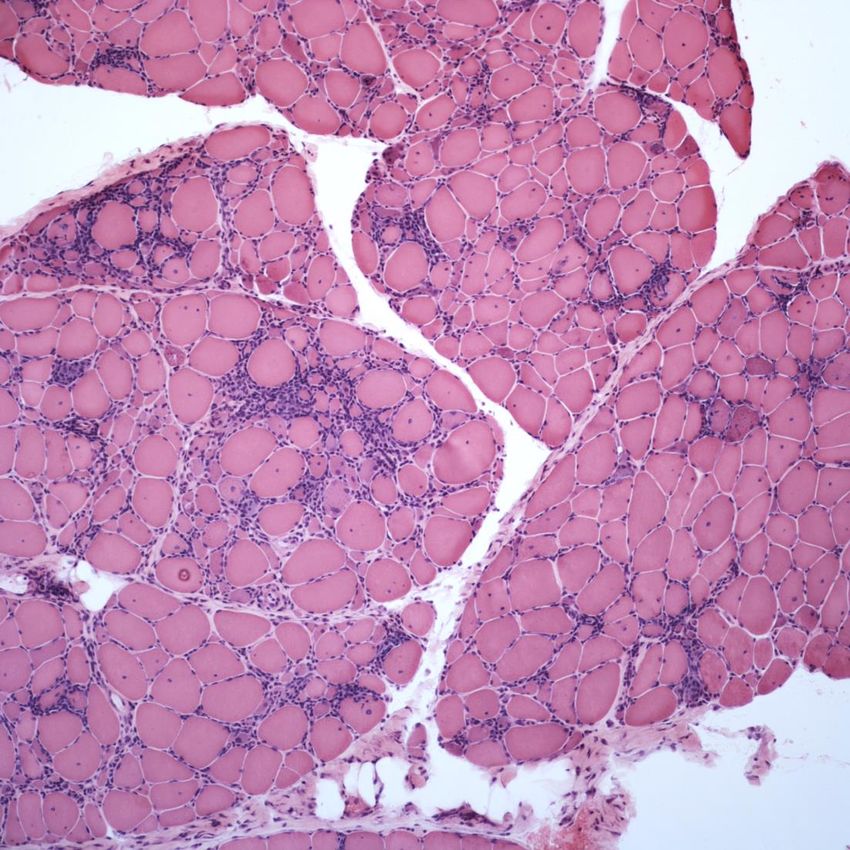
Manifeste Konduktorinnen ca. 10%Dystrophinopathie - Histologie Muskeldystrophes Gewebssyndrom
Muskeldystrophie Typ Duchenne (DMD) • Symptome vor dem 3. LJ beim Erlernen von Stehen/Gehen • CK stark erhöht v.a. im frühen Verlauf • Motorische Entwicklungsverzögerung • Progrediente, proximal betonte myatrophe Paresen • Trendelenburgzeichen („Watschelgang“), Gowers-Manöver • Pseudohypertrophien der (Waden)Muskulatur • Lähmungskoliose und Kontrakturentwicklung (oft 6.-10.LJ) • Progrediente Verschlechterung der Respirationsleistung ab ca. 8.–10. LJ • Obligate Kardiomyopathie mit Arrhythmien • Intelligenzminderung (30-40% IQ < 75) • Tod meist kardiorespiratorisch 2. – 3. Dekade, heute oft längeres Überleben Transition in die Erwachsenen-Neurologie!
Muskeldystrophie Typ Becker (BMD) • Motorische Einschränkung nach dem 8. LJ • Progrediente, proximal betonte myatrophe Paresen • teilw. Lähmungskoliose- und Kontrakturentwicklung • mgl. Verschlechterung der Respirationsleistung • mgl. Kardiomyopathie mit Arrhythmien • CK stark erhöht
Genetik der Dystrophinopathien
Die häufigsten Mutationen, die zu einem
Verlust des ORF führen, sind Deletionen.
Mutationstypen
out-of-frame Deletion/Mutation frame-shift, early stop codon, keine Proteinexpression
instabile mRNA, nonsense-mediated mRNA decay (DMD)
in-frame Deletion/Mutation trunkiertes Protein (BMD)
Therapiekonzept: Exonskipping (AON, zielen auf prä-mRNA und modulieren das Spleißen)
Wandel einer out-of-frame in eine in-frame Mutation (Wiederherstellung des Leserahmens)
Duchenne- zu Becker-PhänotypDMD – Exon Skipping
Proof-of-concept trials:
Exon 51 skipping, Exon 45-52 Del., 13%
Substanz PRO051 (Drisapersen, AON: 2OMePS)
Prosensa (Leiden, NL)/GSK → BioMarin
Substanz AVI-4658 (Eteplirsen, AON: PMO)
AVI BioPharma, UK → Sarepta, USA
Klinische Studien, Stand 05/2017:
BioMarin:
Drisapersen in Phase 3, neg. Ergebnisse
Zulassung 01/16 negativ bewertet (FDA);
Market Authorization Application bei der EMA 05/16 zurückgezogen
Programm 2016 eingestellt
Sarepta Therapeutics:
Eteplirsen in Phase 2b, pos. Ergebnisse
04/16 FDA advisors: negative Bewertung, weitere Daten verlangt
19.09.2016: FDA - accelerated approval (Exondys 51),
Surrogat-Endpunkt Dystrophinexpression im Muskel, nicht Klinik
Confirmatory Study of Eteplirsen in DMD: 4658-301 (PROMOVI), Phase 3
Etabliert, bedingt effektiv: (n=160, 09/14-05/19)
Steroide, Kreatinmonohydrat Seit 01/2017: Antrag auf conditional approval von der CHMP/EMA geprüftDMD - Gentherapie Ataluren (TranslarnaTM): „Stop codon read-through“ Mechanismus, small-molecule compound EU-weite Zulassung zur Behandlung der DMD; seit 12/2014 in Deutschland verfügbar Für folgende Patienten zur Behandlung zugelassen: Patienten mit genetisch gesicherter DMD und dem Nachweis einer sogenannten Nonsense-Mutation (stop codon, ca. 13% aller DMD) und Alter von mindestens 5 Jahren und erhaltener Gehfähigkeit Datenlage: Internationale Phase 2b/3-Studie n=174, 48 Wochen, 2 Dosen von Ataluren (40 mg/kg/d und 80 mg/kg/d), 6MWT Initiale Analysen keine signifikanten Ergebnisse, Subgruppen-Analysen: Gehfähigkeit unter niedrigerer Dosis zu einem geringeren Ausmaß verschlechtert als unter Placebo „Zulassung unter besonderen Bedingungen“ (conditional marketing authorization) in der EU → Daten zu Sicherheit und Wirksamkeit noch zu bestätigen Observational Study/Registry 2015-2022 (n=200)
Myotone Dystrophie Typ 1 (DM1)
Curschmann-Steinert-Erkrankung
Erstbeschreibung:
1909 (Steinert)
1912 (Curschmann)
Prävalenz:
5-15/100.000 (Europa),
Canada (Quebec) 200:100.000
häufigste Form der Muskeldystrophie im Erwachsenenalter,
aufgrund der hohen phänotypischen Variabilität wahrscheinlich stark
unterdiagnostiziertKraniale Manifestationen der DM1 • Stirnglatze, leichte Ptosis • Facies myopathica („myotonica“) • Reduzierte Mimik, fehlende mimische Expression • Temporale Muskelatrophie • Leichte Dysarthrie/Dysphagie
Katarakt bei DM1 • Juvenile Katarakt irisierend, multicolor, posterior, subkapsulär: „Christbaumschmuckkatarakt“, „myotone“ Katarakt; meist junges Erwachsenenalter
Hoher gotischer Gaumen Defekte des Zahnschmelzes, „tented“ upper lip v.a. bei kongenitalen Formen
Multisystemcharakter der DM1
Skelettmuskulatur
Distale Muskelschwäche, Atrophien, Myotonie
Magen-Darm-Trakt
Affektion der glatten Muskulatur,
Mega-Colon, Obstipation,
Pseudo-Obstruktion, CholelithiasisNeuromuskuläre Bildgebung bei DM1 Fettige Degeneration des anterioren > posterioren Oberschenkelkompartiments
Multisystemcharakter der DM1 Hautveränderungen: gehäufte Pilomatrixome Herz Reizleitungsstörungen/Arrhythmien > Kardiomyopathie, maligne Arrhythmien und plötzlicher Herztod fatalste Komplikationen und häufigste Todesursache!
Multisystemcharakter der DM1 Endokrines System Diabetes mellitus (Insulinresistenz), primärer Hypogonadismus, Hodenatrophie, Infertilität, Menstruationsstörungen, hypothalamische Störungen, Schilddrüsenfunktionsstörungen Respiratorisches System Zwerchfell- und Atemmuskelschwäche, obstruktives und zentrales Schlaf-Apnoe-Syndrom, Aspirationspneumonien (bei Dysphagie, häufige Todesursache) Labor CK (oft nur leicht, auch normal), -GT, IgG, Testosteron , IGF-1 , FSH
Zerebrale Beteiligung bei DM1 Fatigue, Tagesmüdigkeit, Hypersomnie!
Zerebrale Beteiligung bei adult-onset DM1
Neuropsychologische Defizite, kognitive Dysfunktion
Störungen der Exekutivfunktionen, kognitive Verlangsamung („slowing“)
Vermeidendes/passives Verhalten
Spez. Persönlichkeitszüge (Apathie, Indifferenz, Antriebsminderung)
„Lack of self-awareness“, „Theory-of-mind“-Probleme
Depression
Kinder: Autismus-Spektrum-Störungen!Zerebrale Bildgebung bei DM1
Marklagerveränderungen (white matter lesions,
besonders typisch: anterior-bitemporale sog. ATWML);
Hirnatrophie, Hyperostose der KalotteBeteiligung der weißen Substanz
Strukturelles MRT (incl. VBM und DTI):
Reduktion der weißen Substanz in allen Hirnregionen (VBM, s.u.)
Ubiquitäre Degradation von Faserverbindungen (DTI)
Corpus callosum, limbisches System und Frontallappen „hot spots“?
Primäre Erkrankung der weißen Substanz?
3.0T-MRI; Minnerop et al. 2011
22 DM1 Patienten vs. KontrollenGenetik der DM1
Beschreibung des Gendefektes 1992
Instabile CTG-Repeat Expansion
in 3` UTR des Dystrophia-Myotonica-Proteinkinase-Gens (DMPK) auf Chromosom 19
Transkribiert, evtl. sogar translatiert: sense und antisense Repeat-assoziierte Non-ATG Translation,
RAN-Translation Proteinpathologie ähnlich HD, SCA8, FXTAS, C9orf72 ALS/FTD
Repeat-Anzahl Erkrankungsalter, -schwere
Instabilität der Repeat-Expansion in Mitose/Meiose
Antizipation (~ 2.9 Dekaden)
Kongenitale Form (maternal)Molekulare Pathogenese der DM1
RNA-Pathologie
Toxic gain-of-function der RNA(CUG)n
nukleäre (und zytoplasmatische) Akkumulation
der expandierten RNA-Transkripte
Bindung und Funktionsstörung von RNA-Bindeproteinen (Spleiß-/Translations-Faktoren MBNL1/2
und CUGBP/CELF1)
Sequestrierung und loss-of-function von MBNL1/2 ()
Hochregulation und gain-of-function von CUGBP/CELF1 ()
„Spleißopathie“: RNA-Instabilität und aberrantes alternatives Spleißen der prä-mRNA anderer Gene
Störung der zellulären Proteinsynthese:
Insulin Rezeptor, CLCN1, CaV1.1 Calcium Kanal, Cardiac Troponin T, RYR-1,
Myotubularin MTM1, SERCA, muscular bridging integrator-1 BIN1, NMDA-Rezeptor 1, APP, TauGentherapie der DM1 Gentherapie-Ziel: Pathogene RNA-Foci (upstream der Pathologie) AON binden an RNA-Expansionen, inhibieren die Bindung von Spleiß-Faktoren oder setzen sie wieder frei; ggf. Degradation der mutierten RNA RNA-Silencing (RNAi/RNA-Interferenz)-Techniken (DMPK knockdown) Gute Erfolge in Zellkultur- und Tiermodellen (Maus, Primaten; dosisabhängige Reduktion von DMPK-Transkripten)
Therapiekonzepte/Studien - DM1 Tideglusib: Glykogensynthase-Kinase-3-Beta (Seronin/Threonin-Kinase, GSK3b)-Inhibitor korrigiert die Aktivität der RNA-Bindeproteine wie CUGBP1 in DM1-Tiermodellen; präklinische Wirksamkeit in transgenen Modellen und DM1 ex vivo Muskelgewebe Klinische Studien: OPTIMISTIC: Kognitive Verhaltenstherapie und körperliches Training, www.optimistic-dm.eu Rekrutierung abgeschlossen, n = 250 IONIS-DMPKRx, Biogen/Ionis, generation 2.5 chimeric AON design “Phase 1/2a blinded, placebo-controlled study to assess the safety, tolerability, and dose-range finding of multiple ascending doses of ISIS 598769 administered s.c. to adult patients with Myotonic Dystrophy Type 1”, n=48, 12/14-11/16 (study completed) 01/2017: “Ionis has decided not to advance its IONIS-DMPK-2.5Rx program”. Neue Technologie für DM1 in Entwicklung. AMO 2, Tideglusib, AMO Pharma Limited “A single-blind, phase 2 study to evaluate the safety and efficacy of Tideglusib 400mg or 1000mg for the treatment of adolescent and adult congenital and juvenile-onset Myotonic Dystrophy Type 1”, 08/16-12/17
Spinale Muskelatrophie
Spinale Muskelatrophie (SMA)
- SMA
Diagnosestellung:
Anamnese, Symptome,
CK-normal/leicht erhöht
→ Genetik: SMN1-Gen
Klinischer Verlauf:
Typ 1: Kein Sitzen
Typ 2: Kein Gehen
Typ 3: Gehverlust
Typ 4: Adulte Form
Therapie:
Symptomatisch,
bisher nicht kausal
Folie 42 TitelSMADefinition
- Genetik
Autosomal rezessiv, Ursache sind pathogene Mutationen
des Survival Motor Neuron 1 (SMN1) Gens auf Chromosom 5
95% der SMA: Exon 7 des SMN1 Gens ist homozygot deletiert
2% der Bevölkerung sind heterozygot (Anlageträger) für eine
SMN1 Deletion (1:50), SMA Inzidenz 1:10.000 Neugeborene
Homologes SMN2 Gen in direkter Nachbarschaft
SMN2 ist im Verlauf der Evolution durch Genduplikationen aus SMN1
entstanden. Die meisten Menschen haben mehrere SMN2 Kopien. SMN2 wird
bei Gesunden nicht benötigt, Genfehler sind häufig. Eine häufige Mutation
führt dazu, dass beim Spleißen Exon 7 übergangen wird. Das dann gebildete
SMN-Protein ist weitgehend funktionslos.
Bei Patienten mit SMA kann SMN2 zum teilweisen Rescue führen
Die Ausprägung einer SMA ist weitgehend davon abhängig,
über wie viele SMN2 Kopien die Betroffenen verfügen
AON Nusinersen verhindert durch Blockade eines repressiven
Genabschnitts,
Folie 43 dass das Exon 7 beim Spleißen übersprungen wird
TitelNusinersen
30.05.2017
Die Europäische Arzneimittel-Agentur (EMA) hat Nusinersen (Spinraza)
für die Behandlung der SMA für alle Verlaufsformen zugelassen
Das Präparat befindet sich seit dem 03.07.2017 im Handel.
FDA-Zulassung seit Ende 2016 für Säuglinge und Erwachsene mit SMA in den USA
Intrathekal appliziert – Tag 1, 14, 30, 60 und danach alle 4 MonateMorbus Pompe
Glykogenose Typ II, Saure Maltase-Mangel,
Glycogen Storage Disease Typ 2 (GSD2)
Mangel an lysosomaler saurer α-1,4-Glukosidase-Aktivität
(„Saure Maltase“, GAA)
Häufigste Glykogenspeicherkrankheit
Geschätzte Inzidenz 1:40.000–1:250.000 Lebendgeburten
Infantile, juvenile/adulte Verlaufsformen (LOPD; u.a. abhängig von
Enzymrestaktivität); große phänotypische Varianz
α-1,4-Glukosidase:
Enzym, das in Lysosomen langkettige Polysaccharide (Glykogen als zu verwertendes Makromolekül)
im Rahmen der Autophagie („Makro-Autophagozytose“) zu Glukose abbaut
Dysfunktion der Autophagie, lysosomale Glykogenakkumulation,
Lysosomen-Ruptur, intrazelluläre GlukoseverarmungM. Pompe
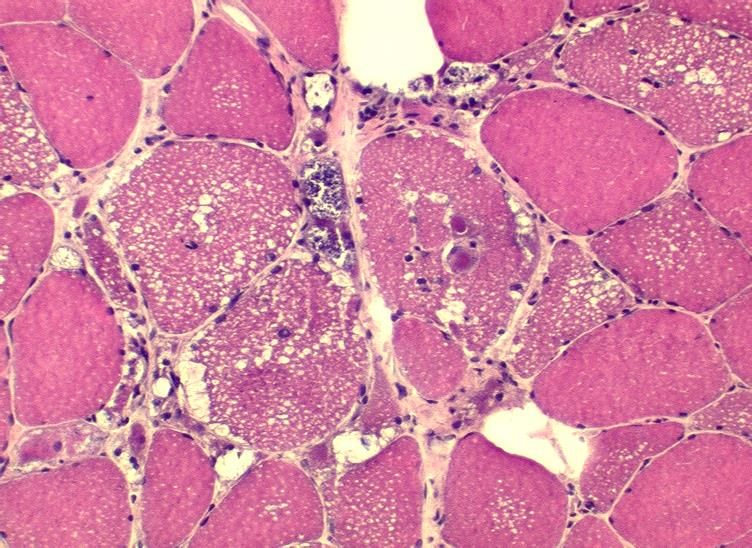
Morbus – Muskelbiopsie
Pompe: Muskelbiopsie
HE; vakuoläre Myopathie
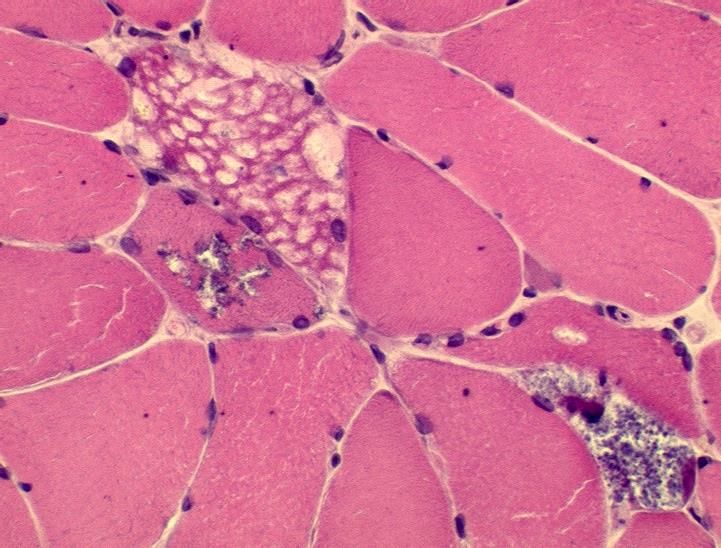
Kann auch unauffällig sein!M. Pompe: Muskelbiopsie PAS-positive Vakuolen; pathologische Glykogenspeicherung
M. Pompe – Infantile Form • Floppy infants • Muskuläre Hypotonie und motorische Retardierung • Hypertrophische Kardiomyopathie mit Rhythmusstörungen • Respiratorische Insuffizienz • Evtl. Hepatopathie • In der Regel Tod im 1. Lebensjahr • Enzymrestaktivität < 1%
Late-onset Morbus Pompe (LOPD)
Klinische Symptomatik variabel, meist Gliedergürtelmuskelschwäche
Beginn sehr variabel, oft 3.-4. Lebensjahrzehnt
Frühe Beteiligung der Atemmuskulatur und Zwerchfellparese
(oft Erstsymptom, paradoxe Atmung)!
Proximal/axial betonte Muskelschwäche (LGMD-Phänotyp, > 80%)
„Skoliose, rigid spine, Kachexie“ als weiterer Phänotyp (ca. 10%)
Selten arterielle Aneurysmata, IOS
Diarrhoen > Stuhlinkontinenz > Urininkontinenz:
häufig, 33-56%
Äußerst selten Herzbeteiligung ( Kindern)
Enzymrestaktivität oft 2-25% (< 30%)M. Pompe
Morbus – Diagnostik
Pompe: Diagnostik
• Anamnese und Klinik
• Muskelenzyme im Serum meist (leicht) erhöht
• EMG: myopathisch, auch SPA
• Biochemie: Enzymaktivitätsbestimmungen, Proteinanalyse
Trockenbluttest als Screening
Biochemie aus Lymphozyten, Fibroblasten, Muskelgewebe
CRIM-Status in Fibroblasten („cross reactive immunological material“,
Western-Blot-Analyse): CRIM-positiv oder-negativ (Kinder)
• Muskelbiopsie bei unklaren Fällen (mit Biochemie)
• Genetik!Kausal orientierte Therapie des Morbus Pompe:
Enzymersatztherapie (ERT)
Alglucosidase Alfa (Myozyme™)
Rekombinant hergestelltes humanes Enzym (rhGAA),
100-kDa Precursor mit Mannose-6-Phosphat (M6P)-Gruppen
M6P-Rezeptor-gebundene Aufnahme über die Zellmembran, intrazelluläres
Verhalten wie endogenes Precursor-Protein
Seit April 2006 durch FDA und EMA zugelassen
Infusionstherapie alle 2 Wochen
Körpergewichts-adaptierte Dosis (20mg/kg KG)Bisherige Ergebnisse der EET
• Erwiesener Nutzen bei Kindern:
überleben z.T. viele Jahre, Ansprechen variabel, andere Probleme können hervortreten
(Dysarthrie, Dysphagie, dilatative statt hypertrophe Kardiomyopathie, ZNS?)
-in Deutschland unter EET mind. 50% verstorben
-mind. 25% aller therapierten Kinder invasiv beatmet
• Bei adulten Formen (LOPD) positive Tendenzen in allen Studien
A), B) LOTS- (open-label extension) Studie (Sanofi-Genzyme);
C), D) eigene Investigator-initiierte Beobachtungsstudie über 12-36 Monate
A), B) n = 90/81, Therapie über 78/104 Wochen:
VK stabil, Gehstrecke (Median) im 6 Minuten-Gehtest
C), D) n = 44/38, Therapie über 12/36 Monate:
VK stabil, Gehstrecke (Median) im 6 Minuten-Gehtest nach 12 und
24 Monaten, wieder leichte Verschlechterung nach 36 Monaten
Review-Arbeit 2016 (J Neurol, Schoser et al.):
22 Studien; 438 Patienten; ~45.7 Monate
“improvements in survival and ambulation, prevention of deterioration in respiratory function”Probleme der EET
Hohe Proteinmenge, teils schwere allergische Reaktionen
Haut, Bronchien, Kreislauf (z.T. erst nach 1-2 Jahren)
Geringe Bioverfügbarkeit in Lysosomen
Im Skelettmuskel Vakuolisierung und Destruktion irreversibel
Autophagie-Problematik, früher Therapiebeginn
Bildung von Antikörpern gegen Alglucosidase Alfa
Neutralisierend? Wirkungsabschwächend? Erhöhtes Risiko für Allergien?
Keine Heilung, Wirkungsreduktion im Verlauf?
Milde Verlaufsformen: Risiko-Nutzen Abwägung
Registry-Programm („natural history“)
Kosten!Entzündliche Myopathien/Myositiden
- Klassifikation -
Polymyositis (PM)
-überdiagnostiziert, nur 2-9% aller entzündlichen Myopathien
-oft Einschlusskörpermyositis, noch öfter Overlap-Myositis
Begleitmyositis bei Kollagenosen = Overlap-Myositis
Dermatomyositis (DM)
Immunvermittelte nekrotisierende Myopathien
(z.B. anti-SRP-, anti-HMGCR-Antikörper)
Sporadische Einschlusskörpermyositis (sIBM)Entzündliche Myopathien/Myositiden
Zusatzbefunde:
• CK: normal oder erhöht (teilweise sehr hoch)
• EMG:
• „irritative Myopathie“
• kleine, kurze, polyphasische Potentiale
• Fibrillationen, positive scharfe Wellen
• Autoantikörper bei 50% aller Patienten mit Myositiden:
• Myositis-spezifische Autoantikörper
in erster Linie bei Pat. mit Myositis (aber nicht ausschließlich)
• Myositis-assoziierte Autoantikörper
meist bei systemischen Immunerkrankungen/Overlap/KollagenosenMyositisches Gewebssyndrom (Polymyositis)
Entzündliche Erkrankung der Skelettmuskulatur durch Autoimmunreaktion gegen intakte
Muskelfasern; vorwiegend T-Zell-vermittelt (zytotoxische T-Zellen gegen unbekanntes Muskel-
Antigen, Invasion nicht-nekrotischer Fasern durch T-Zellen)
Endomysiale und interstitielle Rundzellinfiltrate
FaserinvasionenDermatomyositis (DM) Entzündliche Erkrankung der Skelettmuskulatur durch Autoimmunreaktion gegen muskuläre Kapillaren und interstitielles Bindegewebe (humoral vermittelte Muskelentzündung, CD4- positive T-Zellen, CD20-positive B-Zellen); Ablagerung von MAC an den Muskelkapillaren → Mikroangiopathie → sekundäre Muskelischämie Epidemiologie: Ca. 40% Beginn nach dem 40. LJ, 20-30% im Kindesalter häufigste inflammatorische Myopathie des Kindesalters Assoziation mit Malignomen häufig (v.a. nach dem 50. LJ, v.a. Mamma-/Ovarial-/Bronchial-Karzinome) Assoziation mit anderen Autoimmunerkrankungen sehr selten
Dermatomyositis • Perivaskulär betonte Entzündungsreaktion • Perifaszikuläre Atrophie • MAC-Ablagerung an Muskelkapillaren, intramuskuläre Mikroangiopathie • Kapillarrarefizierung
Dermatomyositis - Klinik
• Proximal betonte Muskelschwäche (in Wochen/Monaten)
• Dysphagie bis zu 50%
• Fieber, Gewichtsverlust gelegentlich
• oft Schwellungen/Überwärmung der Oberarmmuskulatur
• viel Ödem im Muskel-MRT ( Polymyositis)
• Myokarditis: 40% !
• Myositis-spezifische AK bei adulten DM-Patienten:
z. B. Anti-Mi2, Anti-p155/140, Anti-CADM140 (oft amyopathische DM), Anti-PL12
• Hautveränderungen:
• Heliotropfarbenes Erythem: Dekolleté, Gesicht, Augenlider (Ödeme!)
• Gottronzeichen (schuppiges Erythem über Fingerknöcheln)
• Keinig-Zeichen (Mikroaneurysmen und kleine Blutungen an Nagelfalz)
• Subcutane Kalzifikationen (juvenile Dermatomyositis)Therapie immunvermittelter
Myopathien/-sitiden
• Kortikosteroide
• Foudroyante Verläufe: Hochdosis i.v., dann oral
• Sonst langfristig hochdosiert oral, über Monate/Jahre
• Immunsuppressiva
AZT, MTX, Rituximab
MMF, Ciclosporin A, Cyclophosphamid, etc.
• IVIG (auch als add-on) bei schweren und therapierefraktären
Verläufen oder Kontraindikationen gegen Steroide
• Physiotherapie, supportive Maßnahmen
• Tumorbehandlung!Herzlichen Dank
Sie können auch lesen

















































