Endokrinologie - Zomacton
←
→
Transkription von Seiteninhalten
Wenn Ihr Browser die Seite nicht korrekt rendert, bitte, lesen Sie den Inhalt der Seite unten
NEWSLETTER 32/09
Pädiatrische
Endokrinologie
Zur Krise der Endokrinologie 3 – 5
Die Endokrinologie steht im Zeichen der Krise
– vor allem in Ländern wie den USA und Deutsch-
land, wo bereits von Mangelversorgung gespro-
chen wird. Hierzulande darben auch Forschung
und Lehre. Doch manche Länder in Europa zei-
gen, dass es auch anders geht, und so sehen eini-
ge im europäischen Verbund die letzte Chance.
Hyperinsulinämische Hypoglykämie 6 – 10
Die hyperinsulinämische Hypoglykämie entsteht
durch eine gestörte Regulation der Insulinsekreti-
on durch die Betazellen des Pankreas und ist eine
der Hauptursachen hypoglykämischer Hirnschä-
den und mentaler Retardierung. Inzwischen sind
eine Reihe von Mutationen beschrieben worden,
die mit unterschiedlichen Verläufen der Erkran-
kung assoziiert sind. Ein ganz wichtiger Schritt
im Hinblick auf die Therapieoptionen ist die Un-
terscheidung zwischen fokalen und diffusen For-
men.
Silver-Russell-Syndrom 11 – 14
Das SRS zeichnet sich durch klinische wie gene-
tische Heterogenität aus. In den letzten Jahren
wurden große Fortschritte im Verständnis der ge-
netischen wie epigenetischen Veränderungen er-
zielt, die zum SRS führen. Diese lassen vermuten,
dass alle noch so unterschiedlichen (Epi-)Mutati-
onen letztendlich immer das IGF-System beein-
trächtigen.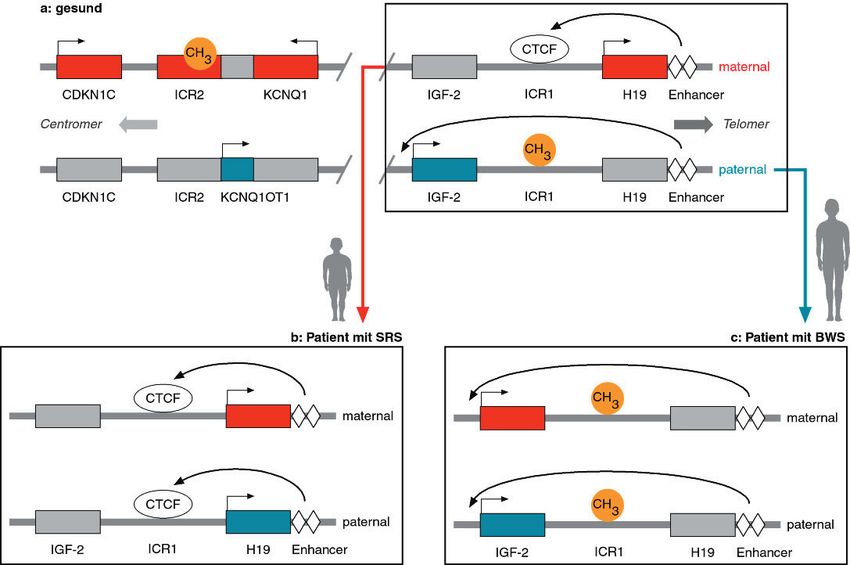
Editorial
25 000 Kinder und Jugendliche in Deutschland leiden an Diabetes. Wegen Zunahme an
Übergewicht und Bewegungsmangel befürchten Experten einen weiteren Anstieg der
Zahlen. „Wir erwarten, dass viele Jugendliche mit Adipositas bald Diabetes Typ 2 bekom-
men“, sagte der Direktor der Leipziger Uniklinik für Kinder und Jugendliche, Professor
Wieland Kiess, im Vorfeld der diesjährigen Jahrestagung der Deutschen Diabetes-Gesell-
schaft (DDG).
Trotz weltweit steigender Krankheitszahlen im Bereich Endokrinologie/Diabetologie
wird diesen Fachbereichen in der Gesundheitspolitik immer weniger Aufmerksamkeit ge-
IMPRESSUM schenkt. Nicht nur der Niedergang der endokrinologischen/diabetologischen Abteilungen
Newsletter Pädiatrische
Endokrinologie in den Kliniken, sondern auch die schrumpfende Zahl niedergelassener Fachärzte spie-
Herausgeber: geln den derzeitigen Zustand der Patientenversorgung wider. Selbst die Kassenärztliche
Ferring Arzneimittel GmbH
Marketing Bundesvereinigung ist bestrebt, den niedergelassenen Fachärzten das endokrinologische
Endokrinologie
Fabrikstraße 7 Labor zu entziehen und nur noch über Laborfachärzte betreiben zu lassen. Diese wieder-
24103 Kiel
Tel.: +49 (0)431 /58 52 –0 um haben in den letzten Jahren eine unbeschreibliche Konsolidierung mitmachen müssen
Fax: +49 (0)431 /58 52– 74
Organisation:
und treten größtenteils nur noch als Großkonzerne auf. Viele sind nicht mehr in deutscher
biomedpark Medien GmbH
Sofienstr. 5 –7
Hand, sondern werden von ausländischen Kapitalanlegern finanziert und gemanagt. Die
69115 Heidelberg
Tel.: +49 (0)6221 /13 747 0
nachhaltige Stärkung der Labormedizin wiederum hat zur Konsequenz, dass Labormedi-
newsletter@biomedpark.de
ziner eigene Ärztezentren unterschiedlicher Fachrichtungen auf- und ausbauen. Tonange-
Fachliche Betreuung:
Dr. Reiner Schmedemann bend sind dann aber nicht mehr die behandelnden Ärzte, sondern die dort eingesetzten
Ferring Arzneimittel GmbH
PD Dr. Klaus Hartmann Verwaltungsleiter. Ähnliches ist auch in vielen Kliniken und Unikliniken geschehen. Der
Praxis für pädiatrische
Endokrinologie Machttransfer geht somit auch nicht spurlos an der Patientenbetreuung vorbei, nach dem
Lyoner Straße 54– 56
60528 Frankfurt Motto „Hauptsache rentabel“. Eine Querfinanzierung „unrentabler“, aber medizinisch
Redaktion:
notwendiger Abteilungen durch „rentable“ Abteilungen gilt oft als unwirtschaftlich. Die
Dr. Corinna Volz-Zang
Gerda Kneifel
Folge ist, dass sich ganze Kliniken oder Abteilungen immer mehr spezialisieren und somit
Lektorat und Layout:
Kirsten Külker nur noch „ihren Teil“ der Patienten betrachten. Dadurch fehlt das – gerade auch für die pä-
Grafik:
diatrische Endokrinologie/Diabetologie unabdingbare – fachübergreifende Qualitätsma-
Erika Heil
art for biomed
nagement. Insbesondere bei der Therapie mit teuren Arzneimitteln kann man hier für den
Burgstr. 70– 74
60389 Frankfurt
klammen Gesundheitsfonds einiges an Geldern einsparen, ohne den Patienten zu schaden.
Copyright:
Copyright und Copyright-
nachweis für alle Beiträge,
Nachdruck, auch auszugs- In diesem Sinne ...
weise, sowie Vervielfälti-
gungen jeder Art nur mit
PD Dr. Klaus Hartmann
schriftlicher Genehmigung
des Verlages
Krise
Zur Krise der Endokrinologie
Mangelversorgung zeichnet sich in vielen Ländern ab
1905 prägte Ernest Starling am Londoner Ro- men wie Schering und Merck haben die Forschung
yal College of Physicians den Begriff Hormon. vorangebracht. In den letzten Jahrzehnten ist die
Nach dieser historischen Vorlesung entwickelte Subspezialität allerdings marginalisiert worden.“ In
sich die Endokrinologie binnen kürzester Zeit zu Deutschland praktizieren bei einer Einwohnerzahl
einem der wichtigsten Zweige der Medizin und von 80 Millionen gerade mal 400 Hormonspezia-
biomedizinischen Wissenschaft. Doch das neue listen in Kliniken und Praxen. 180 Endokrinologen
Jahrtausend steht für dieses interdisziplinäre sind in nicht mehr als 90 Praxen über das Land
Fachgebiet in vielen Ländern im Zeichen einer verteilt [4]. In der Schweiz dagegen kommen laut
Identitätskrise. So debattieren internationale Nieschlag 100 niedergelassene Endokrinologen auf
Endokrinologen bereits seit einigen Jahren über sieben Millionen Einwohner, in Frankreich betreuen
die Zukunft ihrer Subspezialität. nach Angaben der Deutschen Gesellschaft für En-
dokrinologie (DGE) 3000 Endokrinologen 65 Mil-
Weltweit steigender Bedarf lionen Einwohner. Die gravierenden Unterschiede
in der Versorgung bestätigt auch Dr. Ulrich Deuß,
Die Amerikaner haben bereits im Jahr 2003 eine Stu-
Sprecher der Sektion Berufspolitik der DGE. „In
die veröffentlicht, in der sie vor einer gravierenden
Italien ist die Versorgung mit 3 000 Endokrinologen
Unterversorgung im Fachgebiet der Endokrinologie
auf 60 Millionen Einwohner sogar besser als in der
bis zum Jahr 2020 warnten [1]. Im vergangenen Jahr
Schweiz. In Griechenland gibt es allein im Telefon-
rechnete dann Andrew Stewart aus, wie es derzeit
buch von Athen 27 Fachärzte.“ Auch Kinderendo-
um die Versorgung endokrinologischer Patienten
krinologen gibt es in Italien und Spanien deutlich
in den USA steht [2]. Er geht von aktuell 4000 kli-
mehr als in Deutschland. „Allerdings sind nicht
nisch tätigen Endokrinologen in den USA aus, de-
alle in Lohn und Brot“, schränkt Prof. Carl-Joachim
nen 7000 bis 10 000 freie Stellen gegenüberstehen
Partsch vom Endokrinologikum Hamburg ein. Viele
– ohne die Teilzeitstellen zu berücksichtigen. Den
arbeiten quasi ehrenamtlich in Ambulanzen für ihre
zunehmenden Bedarf führt der US-Wissenschaftler
Facharztanerkennung und verdienen sich ihr Geld
auf die zum Teil dramatisch steigenden Prävalenzen
mit Zusatzjobs. In Spanien läuft die Entwicklung der
von Diabetes, Adipositas, Schilddrüsenerkrankun-
Subspezialität der deutschen gerade entgegenge-
gen, Metabolischem Syndrom, Bluthochdruck und
setzt. Hier hat sich die Endokrinologie durch geziel-
Osteoporose zurück. Dass immer mehr Menschen
te Förderung in der jüngsten Vergangenheit stark
einen Hormonspezialisten aufsuchen, erklärt sich
entwickelt. „Noch vor 15 bis 20 Jahren war ihre Be-
nach Stewart auch durch die Aufklärungskampa-
deutung im Land gleich Null“, so Partsch.
gnen bezüglich Diabetes und Adipositas. Hinzu
kommen neue Themenfelder wie Endokrine Dis-
ruptoren, Hormonersatztherapie und Wechseljahre
Mangelnde Honorierung
des Mannes. Die hohe Prävalenz endokriner Erkran- Die Endokrinologie hat ein „Institutionalisierungs-
kungen in den Vereinigten Staaten belegt zudem problem“, das auch andere Subspezialitäten in
eine aktuelle US-Studie aus Baltimore [3]. Hiesige Deutschland haben. Das interdisziplinäre Fachgebiet
Experten gehen davon aus, dass die Entwicklung in wird mehr und mehr auf andere Teilbereiche der
Übersee auf Deutschland übertragbar ist. Medizin aufgeteilt. „Patienten mit Schilddrüsener-
„In Europa müssen wir die Versorgungslage krankungen werden in Deutschland sehr oft von Nu-
allerdings differenziert betrachten“, erläutert Prof. klearmedizinern übernommen. Das ist im Ausland
Eberhard Nieschlag vom Uniklinikum Münster, ge- anders“, nennt Deuß als Beispiel. Auch Kardiologen,
genwärtig Präsident der European Society of Endo- Nephrologen, Gynäkologen und Allgemeinmedizi-
crinology (ESE). „Es gibt gut versorgte Länder, wie ner behandeln Menschen mit endokrinologischen
zum Beispiel Italien, Spanien und Polen. Und es gibt Störungen, doch das ist nicht der einzige Grund für
Länder wie Deutschland, in denen es sehr schlecht die Krise des Fachgebietes. Die Endokrinologie ist
aussieht. Es wundert einen, dass wir so weit zurück- in Deutschland nicht lukrativ, die Verdienstmöglich-
gefallen sind, immerhin waren wir in den zwanziger keiten vergleichsweise gering. Es ist ein ambulantes
bis vierziger Jahren weltweit führend und große Fir- Fach, „fast ohne stationäre Aufgaben – wenn man
3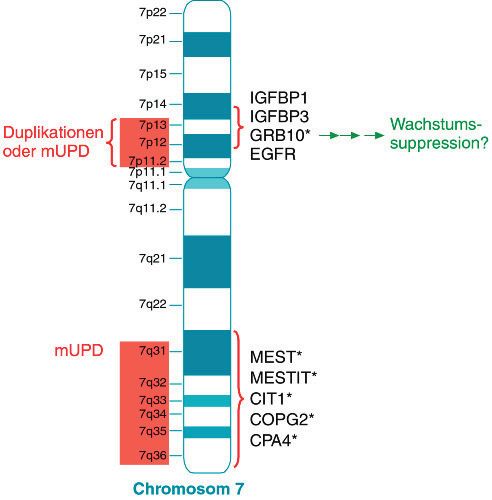
Zukunft
mal vom Drei-Tage-Hunger-Versuch letzten 20 Jahren etwa um die Hälfte geschrumpft,
bei Insulinom absieht oder von Pa- heute sind es noch rund zehn im ganzen Land. Kli-
tienten mit vielen Begleiterkrankun- niken, deren Professoren in Ruhestand gehen, lösen
gen. Behandlungen auf Station sind ihre C4-Lehrstühle wie in Regensburg ganz auf oder
denn auch finanziell nicht abgedeckt sie stufen sie wie in Hannover, Frankfurt oder Marburg
und Unikliniken mit entsprechenden herab und ordnen sie anderen Fachgebieten unter.
Angeboten geraten immer häufiger Diese Entwicklung trifft die pädiatrischen En-
in finanzielle Schwierigkeiten, wie dokrinologen besonders, denn sie sind unter den
die Uniklinik Köln mit ihrer Hormon- ohnehin wenigen Endokrinologen eine Minderheit.
sprechstunde“, weiß Deuß. „In Ulm gab es vor Jahren fünf Kinderendokrinolo-
Doch auch die ambulanten Endo- gen, heute ist es noch einer und auch am Uniklini-
krinologen haben zu kämpfen, noch kum Hamburg-Eppendorf hielt der Klinikleiter die
mehr nach der erneuten Absenkung pädiatrische Endokrinologie für verzichtbar“, zählt
der Regelleistungsvolumina (RLV) im Partsch auf. Ein Lichtblick war die Einrichtung einer
dritten Quartal dieses Jahres. „Für Stiftungsprofessur in Kiel im Jahr 2005. Ob die Pro-
die Beratung ist das Geld schon nach fessur nach Ablauf der Förderung von der Univer-
dem ersten ,Guten Tag, wie geht’s?’ sitätsklinik übernommen wird, bleibt abzuwarten
verbraucht“, formuliert es Deuß be- und wird zumindest von Partsch bezweifelt.
wusst überspitzt. „Für einen Patien- An dieser Situation hat auch die in Deutschland
ten mit Hypophysentumor benötigt sehr spät anerkannte internationale Zusatzausbil-
der Endokrinologe im ersten Quar- dung zum Pädiatrischen Endokrinologen und Dia-
tal zwei bis drei Stunden. Dafür sind betologen offensichtlich nichts geändert. Erst vor
durchschnittlich 30 Euro nicht ausrei- zwei Jahren haben die letzten Bundesländer die
chend, hier wäre vielmehr ein akade- Zusatzausbildung anerkannt – eine im Vergleich
mischer Stundensatz notwendig.“ mit vielen anderen Staaten extrem schleppende
Nach der Abwertung der ärztli- Umsetzung. „Nun haben wir ein Curriculum, aber
chen Beratung und technischer Leis- wo sind die Weiterbildungsstellen?“, fragt Partsch
tungen wie Ultraschall oder Bestim- zudem. „Die Weiterbildung ist de facto nicht finan-
mung der Knochendichte wurden ziert. Darüber hinaus gibt es hierzulande für C4-
dann zu Beginn des Jahres auch die Professoren und Klinikleiter keine Regularien, die
Laborleistungen um ein Fünftel ge- Institutionalisierung einer Subspezialität wie der
senkt. „Der deutsche Endokrinologe Endokrinologie ist damit extrem schwierig.“ Auch
erhält für die Bestimmung eines Hor- Deuß moniert: „Angehende Ärzte sehen während
monwertes nur 40 bis 60 Prozent der ihrer Ausbildung an den Kliniken immer seltener
Erstattung, die sein Schweizer Kol- Patienten mit Hormonstörungen und Praxen kön-
lege für dieselbe Leistung erhält“, nen sich einen Assistenten oft nicht mehr leisten.“
rechnet Partsch vor. „Damit werden
Laborleistungen zur reinen Kostener- Forschung kaum mehr wahrnehmbar
stattung“, klagt auch Deuß, „und die
Wo nicht gelehrt wird, gibt es immer weniger Spezi-
ärztliche Beratungsziffer zum einzigen
alisten, und wo es immer weniger Spezialisten gibt,
Posten für den Endokrinologen.“ Die
wird nicht geforscht. „Eine One-Man-Show kann
Honorierung kann jedoch nicht der
keine Forschung mehr betreiben“, formuliert es
einzige Grund sein, zumal in Ländern
Partsch. „Es wundert mich schon, wie ungünstig wir
wie Italien und Spanien der Verdienst
bei internationalen Veröffentlichungen zum Beispiel
noch niedriger ausfällt.
im Vergleich mit Italien dastehen“, gibt Nieschlag
zu bedenken. „Obwohl dort die Endokrinologen
Abbildung:
Wenige Weiterbildungsangebote oft zwei oder drei Jobs brauchen, um ihre Familie
Nobelpreise mit Bezug zur
Endokrinologie Das geringe Interesse des medizinischen Nachwuch- zu ernähren, und die Laborräume ein ganz anderes
Quelle: mod. nach Wilson JD:
ses dürfte auch im kargen Weiterbildungsangebot Niveau haben als bei uns, sind sie uns in Bezug auf
Clin Endocrinology. 2005 Apr;
62(4): 389-396 begründet liegen. Die Zahl der Lehrstühle ist in den internationale Veröffentlichungen weit voraus. Viel-
4Endokrinologie
leicht wird uns in Deutschland unser Perfektionismus ren. Als europäische Gesellschaft finden wir langsam
zum Verhängnis. Wir fangen erst an zu arbeiten, wenn Gehör.“ Eine der ersten Maßnahmen der europäi-
das Drumherum perfekt ist.“ Partsch drückt es noch schen Endokrinologen ist die Formulierung inter-
deutlicher aus: „Es ist doch eine Peinlichkeit, dass nationaler Richtlinien für Diagnose und Therapie
wir, nachdem wir lange Zeit weltweit führend waren zu allen endokrinologischen Krankheitsbildern. Zu
in der Endokrinologie, heute selbst kleinen Ländern Cushing-Syndrom und Nebennierenerkrankungen
wie den Niederlanden hinterherhinken. Wir publizie- beispielsweise hat die ESE bereits gemeinsam mit
ren heute mit Mühe und Not so viel wie die Schwe- der US-amerikanischen Society for Endocrinology
den mit ihren neun Millionen Einwohnern.“ Bestä- Richtlinien publiziert. „Zur Fettsucht und auch zum
tigt werden diese Einschätzungen durch einen Blick Adrenogenitalen Syndrom wird in den kommenden
auf die Deutsche Forschungsgemeinschaft (DFG). drei Monaten etwas veröffentlicht. Darüber hinaus
Schon im Jahr 2003 kamen weniger als zwei Prozent sind etwa zehn weitere Richtlinien in Arbeit.“
aller DFG-Förderungen rein endokrinologischen
Themen zugute, etwas mehr als drei Prozent zielten Kooperationen als Notlösung
auf die Volkskrankheit Diabetes. Entsprechend spielt
In wirtschaftlich schwierigen Zeiten rückt man en-
die deutsche Endokrinologie auf internationalem Ni-
ger zusammen. Zum einen sind durch die Ände-
veau nur noch eine untergeordnete Rolle.
rung des Vertragsarztrechtsänderungsgesetzes
neue Kooperationen möglich geworden. Zum
Rettung im europäischen Verbund? anderen zeichnet sich in der Laborlandschaft ein
Die Wirtschaftskrise macht die Sache nicht besser. Trend zu Fusionen und Großlaboren ab. „Bislang
„Unter den jetzigen wirtschaftlichen Bedingungen kann aber das Labor nur schwer rationalisiert wer-
der Länder sehe ich die Zukunft der Endokrinologie den, weil eine persönliche Anwesenheitspflicht
und die Ausbildung des Nachwuchses an den Uni- des Arztes besteht, wenn er Laborwerte bestim-
kliniken noch schwärzer als in den letzten Jahren“, men lassen möchte. Er darf seine Praxis also nicht
schätzt Partsch die Lage ein. „Endokrinologen be- zwei Straßen weiter haben“, berichtet Deuß. „Wir
klagen sich seit zehn Jahren über ihre schlechte haben einen Antrag laufen, der es möglich machen
finanzielle Lage, in der Hoffnung, dass die Politi- könnte, Labore auch dann gemeinsam zu nutzen,
ker aufwachen. Gezielte Unterstützungsprogram- wenn man nicht im selben Gebäude sitzt. Danach
me wären notwendig, doch das kostet Geld. Und könnten in einem Labor Proben aus unterschiedli-
unter dem Strich ist selbst ohne Krise nichts pas- chen Städten untersucht werden – sofern die Pro-
siert.“ Etwas optimistischer äußert sich Deuß: „Ich ben ordnungsgemäß geliefert und die Apparatu-
Literatur bin guter Dinge, dass in den kommenden Jahren ren sachgemäß bedient werden. Der behandelnde
etwas passieren wird, wenn sich die Versorgung Arzt kann dann online die Messung kontrollieren,
1. Rizza R et al.: A Model
to Determine Workforce weiter verschlechtert hat. In Nordrhein-Westfalen die Werte auf ihre Schlüssigkeit hin kontrollieren
Needs for Endocrinologists zum Beispiel gab es vor Kurzem eine Aussprache und prüfen, ob das Ergebnis zu dem individuellen
in the United States Until
2020. J Clin Endocrinol Me-
im Landtag, in der lokale Politiker bessere Vergü- Patienten passt.“ Sollte die Kassenärztliche Verei-
tab. 2003; 88(5): 1979-1987 tungen gefordert haben. In diesem Bundesland nigung Nordrhein den Antrag genehmigen, „wird
2. Stewart A: The United sind die Regelleistungsvolumina im bundesweiten es ein Mustermodell werden, das sicher viele nach-
States Endocrinology Work-
Durchschnitt sehr niedrig und die Landespolitiker ahmen werden“. Partsch steht Großlaboren skep-
force: A Supply-Demand
Mismatch. J Clin Endocrinol merken langsam, dass ihre Region leidet, auch tisch gegenüber: „Dann rechnen sich die Labore
Metab. 2008 Apr; 93(4): wenn sie sich noch nicht durchsetzen können. Noch nur noch über die Masse, seltenere Hormone und
1164-1166
werden die Schrauben enger gedreht, aber in fünf die Diagnostik seltener Krankheiten werden nicht
3. Golden SH et al.: Clinical
review: Prevalence and bis zehn Jahren wird das alte System neue Grenzen mehr abgedeckt.“ Das Problem sieht auch die
incidence of endocrine and erhalten“, ist sich der Kölner Endokrinologe sicher. DGE: Jeder fünfte Patient mit Hormonstörungen
metabolic disorders in the
Nieschlag sieht die Rettung im europäischen leidet an einer selteneren Erkrankung. „Gerade
United States: a comprehen-
sive review. J Clin Endocri- Verbund. „Gemeinsam haben wir mehr Einfluss und diese Patienten bedürfen einer zeitaufwendigen
nol Metab. 2009 Jun; 94(6): können Politiker und Funktionäre sensibilisieren. Wir Beratung und hochwertigen Diagnostik im Labor“,
1853-1878
arbeiten eng mit dem European Board of Endocri- so Deuß. Insbesondere für sie entwickeln sich die
4. Pressemitteilung DGE
vom 6. März 2009, nology (UEMS) zusammen und versuchen auch mit Dinge derzeit nicht günstig.
www.endokrinologie.net den amerikanischen Endokrinologen zu kooperie- Gerda Kneifel
5HH
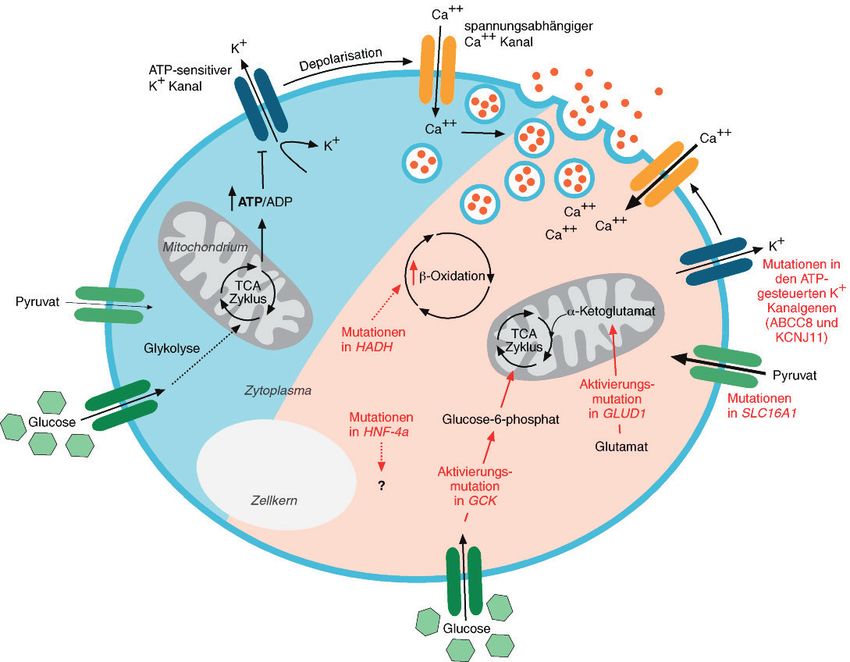
Die hyperinsulinämische Hypoglykämie
Die hyperinsulinämische Hypoglykämie (HH) ist Die kongenitale HH und die sie
die Folge einer unangemessenen und unregu- verursachenden Mutationen
lierten Sekretion von Insulin aus pankreatischen
Die kongenitale HH ist im Hinblick auf Histologie,
Betazellen. Diese Hypersekretion führt zur
molekulare Biologie und Genetik eine sehr hete-
Aufnahme von Glukose durch insulinsensitive
rogene Erkrankung. Es gibt sowohl sporadische
Gewebe – vor allem Skelettmuskel, Fettgewe-
als auch familiäre Formen der angeborenen HH,
be und Leber – und zur gleichzeitigen Hem-
wobei die sporadischen Formen sehr selten sind
mung von Glykogenolyse, Glukoneogenese,
(1:40 000 bis 50 000 Lebendgeborene), die fa-
Lipolyse und Ketolyse. Die dadurch bedingte
miliäre Form dagegen mit einer Inzidenz von bis
Hypoglykämie kann im Nüchternzustand, nach
zu 1:2500 in ethnischen Gruppen mit hoher Kon-
Anstrengung oder postprandial aufgrund einer
sanguinität deutlich häufiger auftritt. Die histolo-
proteinreichen Mahlzeit auftreten. Die hyperin-
gische Differenzierung der kongenitalen hyperin-
sulinämische Hypoglykämie ist eine der Haupt-
sulinämischen Hypoglykämie in fokale und diffuse
ursachen von mentalen Retardierungen und
Formen war ein ganz zentraler Wendepunkt in der
Hirnschäden [1].
chirurgischen Behandlung dieser Erkrankung [3].
Es gibt verschiedene Ursachen der hyperinsuli- Inzwischen wurden sieben verschiedene Gene be-
nämischen Hypoglykämie, die zudem mit sehr schrieben, bei denen Mutationen zu einer fehlre-
unterschiedlichen Erscheinungsformen assoziiert gulierten Insulinsekretion führen (siehe Tabelle 1).
sind. In Tabelle 1 sind die Differenzialdiagnosen Die bisher beschriebenen Mutationen betreffen
der hyperinsulinämischen Hypoglykämie aufge- Gene, die die Schlüsselkomponenten der Insulin-
führt. Auch sind eine Reihe von Syndromen mit sekretion in den Betazellen des Pankreas regulie-
diesem Krankheitsbild assoziiert (siehe Tabelle 2). ren (siehe Abbildung 1) [3].
Das Beckwith-Wiedemann-Syndrom (BWS) ist das
Syndrom, das am häufigsten mit HH in Verbindung KCNJ11 und ABCC8-Mutationen
gebracht wird. Die Inzidenz des Hyperinsulinismus Die glukoseinduzierte Insulinsekretion in den Be-
liegt bei Kindern mit BWS bei etwa 50 Prozent [2]. tazellen wird teilweise durch ATP-sensitive Kalium-
Die HH kann vorübergehend sein. In diesen Fäl- kanäle (KATP) reguliert. Diese Kanäle setzen sich aus
len ist sie meist asymptomatisch und verschwindet der Kalium-Kanaluntereinheit Kir6.2, die von dem
innerhalb der ersten Lebenstage. Zwar sind noch Gen KCNJ11 kodiert wird, und dem Sulfonylharn-
nicht für alle Syndrome die molekularen Mecha- stoff-Rezeptor 1 (SUR1) zusammen, der von dem
nismen der Entstehung der HH bekannt. Beim Gen ABCC8 kodiert wird. Der Glukose-Metabolis-
Beckwith-Wiedemann-Syndrom wird jedoch eine mus in den Betazellen führt zu einer Verschiebung
Überexpression von IGF-2 (Insulinähnlicher Wachs- des intrazellulären ATP:ADP-Verhältnisses in Rich-
tumsfaktor II) aufgrund der Hypermethylierung des tung ATP. Durch diese Verschiebung wird der Ka-
väterlichen UDP11- oder H19-Gens als Erklärung liumkanal geschlossen, die Zelle depolarisiert und
herangezogen. Die Überexpression von IGF-2 durch die Depolarisation öffnen sich spannungs-
könnte durch den wachstumsfördernden Effekt abhängige Calcium-Kanäle. Ca2+ strömt in die
zu einer Hyperplasie pankreatischer Betazellen Zelle ein und vermittelt die Sekretion des Insulins.
führen [2]. In der Publikation „Hyperinsulinism in Sowohl für KCNJ11 als auch für ABCC8 sind Mu-
Developmental Syndromes“ befassen sich Ritika tationen beschrieben worden, die zur HH führen.
Kapoor et al. ausführlich mit den Syndromen, die Beide Gene sind auf dem Chromosom 11p15.1
mit Hyperinsulinismus assoziiert sind und beschrei- lokalisiert. Rezessive inaktivierende Mutationen in
ben die bisher gefundenen molekularen Ursachen den beiden Genen führen zu den häufigsten und
[2]. Aus Platzgründen kann an dieser Stelle nicht schwersten Formen der kongenitalen HH. Pati-
ausführlicher darauf eingegangen werden. enten mit Mutationen in diesen Genen sprechen
6HH
normalerweise nicht auf eine medikamentöse The- HFN4A-Mutationen
rapie an und benötigen eine Pankreatektomie. Da- Das Gen HFN4A kodiert den Hepatozyten-Nukle-
gegen führen autosomal-dominante Mutationen zu ären-Faktor-4∙, einen Transkriptionsfaktor, der Teil
einer milderen, behandelbaren Form. Mutationen eines Regulationsnetzwerkes aus Transkriptionsfak-
in den Genen KCNJ11 und ABCC8 machen etwa toren ist, die die Genexpression regulieren [5]. Es
50 Prozent der kongenitalen HH aus, in manchen ist bekannt, dass Mutationen im HFN4A-Gen den
Populationen wie den Japanern sind es dagegen MODY-Diabetes (maturity-onset diabetes of young)
nur 20 Prozent [4]. verursachen, der durch autosomal-dominante Ver-
erbung und eine eingeschränkte glukoseinduzierte
GLUD1- und GCK-Mutationen
Insulinsekretion aus Pankreaszellen gekennzeichnet
Die aktivierte Glutamat-Dehydrogenase führt zu
ist. Inzwischen wurden heterozygote Mutationen
einer verstärkten Oxidation von Glutamat und er-
in HNF4A beschrieben, die mit vorübergehender
höht so das ATP :ADP-Verhältnis in den Betazellen,
oder dauerhafter HH sowie Makrosomie assoziiert
wodurch die Insulinfreisetzung gesteigert wird (sie-
sind [6]. In Mausmodellen wurde eine 60-prozen-
he oben). Normalerweise wird das Enzym durch
tige Reduktion der Expression des Proteins Kir6.2
GTP gehemmt und durch Leucin aktiviert. Aktivie-
nachgewiesen, das – wie oben beschrieben – Teil
rende Mutationen im Glutamat-Dehydrogenase-
des Kaliumkanals ist [7]. Allerdings wurden in ande-
Gen GLUD1 schwächen den inhibitorischen Effekt
ren Studien keine Expressionsänderungen des ent-
von GTP ab und erleichtern die Aktivierung durch
sprechenden Gens KCNJ11 nachgewiesen, sodass
Leucin. Die Glukokinase katalysiert in Pankreaszel-
der Mechanismus noch immer unklar ist.
len beim Eintritt in die Glykolyse die Phosphory-
lierung von Glukose und damit den geschwindig- HADH-Mutationen
Tabelle 1: keitsbestimmenden Schritt im Metabolismus der Das mitochondriale Enzym HADH (Hydroxyacyl-
Differenzialdiagnosen
Glukose. Aktivierende Mutationen im Gen für die CoA-Dehydrogenase) katalysiert den vorletzten
der hyperinsulinämischen
Hypoglykämie Glukokinase (GCK) senken den Schwellenwert der Schritt in der ‚-Oxidation der Fettsäuren. Dabei
Quelle: modifiziert nach [3] glukosestimulierten Insulinsekretion. entsteht 3-Ketoacyl-CoA. Mutationen, die zu ei-
nem Funktionsverlust des HADH-Gens führen,
Kongenitale hyperinsulinämische Hypoglykämie verursachen eine kongenitale HH, wobei auch hier
• autosomal rezessive Mutationen: ABCC8, KCNJ11, HADH
• autosomal dominante Mutationen: ABCC8, KCNJ11, GCK, GLUD1, HNF4A, SLC16A1
bisher die molekularen Mechanismen nicht aufge-
klärt sind. Es scheint jedoch so zu sein, dass HADH
Syndrome, die mit HH assoziiert sind (siehe Tabelle 2)
mit anderen Genen interagiert, die für die Beta-
Postprandiale hyperinsulinämische Hypoglykämie zellentwicklung und -funktion wichtig sind [8].
• Dumping-Syndrom
• nach gastrischem Bypass bei adipösen Erwachsenen SLC16A1-Mutationen – anstrengungsinduzierte HH
• pankreatogene hyperinsulinämische Non-Insulinoma-Hypoglykämie
• Mutationen im Gen für den Insulinrezeptor Bei der anstrengungsinduzierten hyperinsulin-
• Insulin-Autoimmunsyndrom ämischen Hypoglykämie setzt die Hypoglykämie
Insulinom gewöhnlich etwa 30 Minuten nach einer kurzen
• sporadisches Insulinom Phase anaerober körperlicher Betätigung ein. Die-
• multiple endokrine Neoplasie Typ 1
se Form der Hypoglykämie entsteht durch Mutati-
metabolische Ursachen
onen in der Promotorregion des SLC16A1-Gens,
• kongenitale Störungen der Glykosylierung (Typ 1a, 1b und 1d)
• Tyrosinämie Typ 1 die zu einer verstärkten Expression des Monocarb-
oxylat-Transporters 1 (MCT1) führt. Normalerweise
künstliche hyperinsulinämische Hypoglykämie
ist die Expression von MCT1 in Pankreaszellen sehr
andere Ursachen (meist vorübergehend) gering, wodurch der Einfluss von Pyruvat und Lak-
• mütterlicher Diabetes mellitus (Gestationsdiabetes und insulinabhängiger Diabetes)
• intrauterine Wachstumsverzögerung (SGA, Small for Gestational Age) tat auf die Insulinsekretion niedrig gehalten wird.
• perinatale Asphyxie Die erhöhten Spiegel von MCT1 bei dieser Form
• Rhesus-Isoimmunisierung
der Hypoglykämie ermöglichen die verstärkte Sti-
7HH
und Kollegen stellen in ihrer Über-
Beckwith-Wiedemann-Syndrom
prä- und postnatales übermäßiges Wachstum Sotos-Syndrom sichtsarbeit einige Befunde dar, auf
Simpson-Golabi-Behmel-Syndrom die hier im Einzelnen jedoch nicht
Kabuki-Syndrom eingegangen werden kann [3].
postnatale Wachstumsstörungs-Syndrome
Costello-Syndrom
Trisomie 13 (Patau-Syndrom)
Syndrome mit chromosomalen Auffälligkeiten
Mosaikform des Turner-Syndroms
Klinik
Syndrome, die zu Störungen der Die hyperinsulinämische Hyperglyk-
Timothy-Syndrom
Calcium-Homöostase führen (intermittierende HH) ämie kann ganz verschiedene Ur-
an das ABCC8-Gen angrenzende Gendeletion, sachen haben. Entsprechend
Usher-Syndrom
die das ABCC8-Gen beeinflusst
unterschiedlich können Alter bei Er-
angeborene Störung der Glykosylierung 1a
krankungsbeginn, Schweregrad und
angeborene Störungen der Glykosylierung angeborene Störung der Glykosylierung 1b
angeborene Störung der Glykosylierung 1c Krankheitsverlauf sein. Die schweren
Formen manifestieren sich bereits
Tabelle 2: mulation des mitochondrialen Metabolismus durch in der Neonatalperiode. Die Symptome können
Syndrome, die mit einer
hyperinsulinämischen
Pyruvat oder Laktat [9]. unspezifisch sein – wie Ernährungsprobleme, Le-
Hypoglykämie (HH) asso- thargie und Irritabilität oder auch spezifische Sym-
ziiert sind Andere Formen der HH ptome wie Apnoe, Krampfanfälle oder Koma. Die
Quelle: modifiziert nach [2]
Während für die kongenitale HH eine Reihe von Makrosomie, ein häufiges Merkmal der kongenita-
Mutationen beschrieben wurden, sind für die an- len hyperinsulinämischen Hypoglykämie im Neu-
deren Formen der HH, wie die postprandiale HH, geborenenalter, ist die Folge der fetalen Hyperin-
die Ursachen meist nicht aufgeklärt. Ritika Kapoor sulinämie. Leichtere Formen der HH können auch
Abbildung 1:
Die an der Regulation der
Insulinsekretion beteilig-
ten molekularen Mecha-
nismen sind vereinfacht im
blauen Bereich dargestellt,
im rosa Bereich die bei
HH nachgewiesenen
Mutationen, die auf diese
Regulation einwirken
Die verschiedenen Mutati-
onen beeinflussen sowohl
direkt den Glukosestoff-
wechsel als auch verschie-
dene Transporter.
Quelle: modifiziert nach [3]
8HH
erst im späten Kleinkind- oder Kindesalter auftre- xie kann entweder zu einer vorübergehenden oder
ten und durch wiederholte Symptome der Hypo- anhaltenden HH führen und eine Behandlung mit
glykämie deutlich werden. Eine perinatale Asphy- Diazoxid notwendig machen [10].
Diagnose der kongenitalen HH
• zur Aufrechterhaltung der Normoglykämie Glukoseinfusionsrate > 8mg /kg /min erforderlich (normaler Bereich
4 – 6mg /kg /min)
• Blutglukosespiegel < 3mmol /l
• unter Hypoglykämie nachweisbar zu hohe Insulinspiegel und/oder erhöhte Spiegel vom C-Peptid bei gleichzeitig
nicht nachweisbaren Ketonkörpern und Fettsäuren, die den metabolischen Effekt von Insulin widerspiegeln
• bei Kindern mit Hyperinsulinismus und Hyperammoniumämie sind die Plasma-Ammoniak-Spiegel erhöht
• positive glykämische Reaktion auf Glukagon
• auffällige Acylcarnitinspiegel und /oder
• Hypoglykämie nach Protein- oder Leucinbeladung
• Hypoglykämie nach Belastungstest oder Pyruvatbeladung
Diazoxidtherapie
positive Therapieresponse keine Therapieresponse
Überprüfung der Nüchterntoleranz Screening auf ABCC8- und KCNJ11-Mutationen
väterliche ABCC8- und/oder Homozygotie oder gemischte Hetero-
KCNJ11-Mutationen zygotie für ABCC8 und/oder KCNJ11
(hochindikativ für fokale Läsionen) (Patient erbt zwei rezessive Allele – ent-
oder keine Mutationen weder für ABCC8 oder KCNJ11, die
in ABCC8/KCNJ11 eine kongenitale HH in einem heterozy-
(fokale Läsionen unwahrscheinlich) goten Status verursachen können)
18
F-L-Dopa-PET-CT-Scan
diffuse Erkrankung
fokale Erkrankung
• hochkalorische Diät und/oder häufige Nahrungs-
aufnahme
chirurgische Resektion der fokalen Läsion
• Octreoidtherapie
(vorzugsweise mit
• fast vollständige Pankreatektomie
laparoskopischem Eingriff)
Abbildung 2:
Diagnose- und Therapie-
algorithmus der kongeni-
Follow-up:
talen hyperinsulinämischen • Kontrolle von Wachstum und Entwicklung
Hypoglykämie • neurologische Kontrollen
• genetische Beratung
Der 18F-L-Dopa-PET-CT-
Scan ist nur bei Patienten nach einer annähernd totalen Pankreatektomie:
indiziert, die potenziell eine
• Behandlung des Diabetes mellitus
fokale Läsion haben. • Kontrolle der exokrinen Funktionen des Pankreas
Quelle: modifiziert nach [2]
9HH
5. Lucas B: Funktion des Beginnt die HH erst im Erwachsenenalter, so fusen kongenitalen HH wirksam ist – mit Ausnah-
zellspezifischen Transkripti-
onsfaktors HNF4∙ ∙ bei der handelt es sich in den meisten Fällen um ein Insuli- me der autosomal rezessiven Mutationen in den
Zellproliferation und Identifi- nom. Das mittlere Alter bei Diagnosestellung ist um ABCC8- und KCNJ11-Genen [3]. Einige Patienten
zierung von HNF4∙-regulier-
ten Genen in Nierenzellen. die 45 Jahre. Bei Patienten, die ein Insulinom in As- mit Mutationen im CGK-Gen benötigen eine sub-
2005. Inaugural-Dissertation. soziation mit einer multiplen endokrinen Neoplasie totale Pankreatektomie [12]. In Abbildung 2 sind
http://deposit.ddb.de/cgi-
bin/dokserv?idn=976825031
Typ 1 haben, tritt die Krankheit meist in einem Alter Diagnose- und Therapiealgorithmus für Patienten
&dok_var=d1&dok_ext=pdf& um die 25 Jahre auf. Bei Erwachsenen lassen sich die mit kongenitaler hyperinsulinämischer Hypoglykä-
filename=976825031.pdf
Symptome der HH zum einen in solche unterteilen, mie dargestellt.
6. Pearson ER et al.: Macro-
somia and hyperinsulinaemic die durch hypoglykämiebedingte Entladungen im Die Unterscheidung zwischen fokaler und diffu-
hypoglycaemia in patients autonomen Nervensystem verursacht werden. Dazu ser kongenitaler HH hat für die Therapie insofern
with heterozygous mutations
in the HNF4A gene. PLoS gehören Hunger, Schweißausbrüche, Parästhesie, deutliche Fortschritte gebracht, als Patienten mit
Med. 2007 Apr; 4(4): e118 Angst, Tremor und Herzrasen. Zum anderen kann der fokaler Erkrankungsform durch partielle Pankreat-
7. Gupta RK et al.: The
MODY1 gene HNF-4alpha
Glukosemangel im Gehirn zu neuroglykopenischen ektomie vollständig geheilt werden können. Eine
regulates selected genes Symptomen wie Verhaltensänderungen, Konfusion, ganz wichtige Rolle spielen hierbei F-L-DOPA-
18
involved in insulin secretion.
J Clin Invest. 2005 Apr;
Lethargie, Sehstörungen, Persönlichkeitsverände- PET-Untersuchungen, die inzwischen eine akku-
115(4): 1006-1015 rungen, Krämpfen und Bewusstlosigkeit führen [11]. rate präoperative Lokalisation fokaler Läsionen
8. Lantz KA et al.: Foxa2 re-
des Pankreas erlauben. Sie beruhen darauf, dass
gulates multiple pathways of
insulin secretion. J Clin Invest. Diagnose L-DOPA von den Inselzellen des Pankreas aufge-
2004 Aug; 114(4): 512-520 nommen und durch die artomatische Aminosäu-
9. Ishihara H et al.: Overex- Der wichtigste biochemische Hinweis auf eine un-
pression of monocarboxy- redecarboxylase zu Dopamin umgewandelt wird.
regulierte Insulinsekretion ist der erhöhte Glukose-
late transporter and lactate Die Aufnahme des Positronen-emittierenden 18F-L-
dehydrogenase alters insulin bedarf zur Aufrechterhaltung einer Normoglykämie
secretory responses to
DOPA ist in Betazellen mit einer erhöhten Bildung
(> 8mg /kg /min, Normalbereich 4–6mg/kg/min).
pyruvate and lactate in beta und Sekretion von Insulin im Vergleich zu nicht be-
cells. J Clin Invest. 1999 Diagnostiziert wird die HH durch den Nachweis ei-
troffenen Zellen erhöht [13]. Während früher die
Dec; 104(11): 1621-1629 ner unangemessenen Insulinfreisetzung (und/oder
10. Hoe FM et al.: Clinical partielle und subtotale Pankreatektomie als offene
unangemessene C-Peptidspiegel) während einer
features and insulin regulati- Operation durchgeführt wurde – mit den entspre-
on in infants with a syndrome hypoglykämischen Phase [3]. Bei Erwachsenen mit
of prolonged neonatal hy- chenden peri- und postoperativen Komplikationen
HH ist zudem die Bestimmung von Insulinantikör-
perinsulinism. J Pediatr. 2006 – wird inzwischen bei fokalen Formen der Eingriff
Feb; 148(2): 207-212 pern notwendig, um ein Insulin-Autoimmunsyn-
11. DeRosa MA et al.: Hypo-
teilweise auch laparoskopisch vorgenommen [3].
drom auszuschließen.
glycemia and the sympatho- Indem immer mehr Informationen über die ge-
adrenal system: neurogenic
netischen Ursachen der HH gewonnen werden, las-
symptoms are largely the Therapie
result of sympathetic neural, sen sich heute für einige Formen bereits ziemlich
rather than adrenomedulla- Die frühzeitige Diagnose, die Vermeidung hypo-
ry, activation. Am J Physiol
präzise der Verlauf und Schweregrad vorhersagen,
Endocrinol Metab. 2004 Jul; glykämischer Episoden sowie die sofortige Be- was wichtige Implikationen für die Therapie hat.
287(1): E(e)32-41 handlung etwaiger Hypoglykämien sind die Eck-
12. Glaser B, Hirsch HJ, Dr. Corinna Volz-Zang
Landau H: Persistent hyper-
pfeiler der Behandlung der kongenitalen HH, um
Literatur
insulinemic hypoglycemia Hirnschäden und mentale Retardierung zu verhin-
1. Menni F et al.: Neurologic outcomes of 90 neonates and in-
of infancy: long-term octreo-
dern [3]. Die Hauptstütze der Therapie ist die Be- fants with persistent hyperinsulinemic hypoglycemia. Pedia-
tide treatment without pan-
trics. 2001 Mar; 107(3): 476-479
createctomy. J Pediatr. 1993 reitstellung großer Mengen von Kohlenhydraten,
Oct; 123(4): 644-650 2. Kapoor RR et al.: Advances in the diagnosis and management
um die Normoglykämie aufrechtzuerhalten. Dies of hyperinsulinemic hypoglycaemia. Nat Clin Pract Endocrinol
13. de Lonlay P et al.:
Congenital hyperinsulinism: ist in vielen Fällen nur durch einen zentralvenösen Metab. 2009 Feb; 5(2): 101-112. Review
pancreatic [18F]fluoro-L- Katheter möglich, über den die Zufuhr konzen- 3. Kapoor RR et al.: Hyperinsulinism in developmental syndro-
dihydroxyphenylalanine mes. Endocr Dev. 2009; 14: 95-113
(DOPA) positron emission trierter Dextroselösung sowie das Monitoring der
4. Tanizawa Y et al.: Genetic analysis of Japanese patients with
tomography and immu- Blutglukosespiegel in kurzen Zeitabständen mög- persistent hyperinsulinemic hypoglycemia of infancy: nucleo-
nohistochemistry study of tide-binding fold-2 mutation impairs cooperative binding of
DOPA decarboxylase and lich ist. Die medikamentöse Therapie umfasst die
adenine nucleotides to sulfonylurea receptor 1. Diabetes. 2000
insulin secretion. J Clin En- Gabe von Diazoxid, eines Antagonisten des KATP- Jan; 49(1): 114-120
docrinol Metab. 2006 Mar;
91(3): 933-940 Kanals, der in der Regel bei allen Formen der dif- Fortsetzung Literatur siehe linke Spalte
10S RS
Das Silver-Russell-Syndrom
Epigenetische Veränderungen als Ursache für Heterogenität
Epigenetik, genomisches Imprinting und uniparentale Disomie
In Säugetieren werden autosomale Gene typischerweise von beiden elterlichen Allelen exprimiert. Seit Mitte der
80er Jahre ist jedoch bekannt, dass die Expression einer Reihe von mittlerweile etwa 80 Genen von ihrem elter-
lichen Ursprung abhängig ist [a]. Die Epigenetik beschäftigt sich mit diesen elterlich bedingten Modifikationen
der Genexpression, die als Prägung oder Imprinting bezeichnet werden. Im Gegensatz zu DNA-Mutationen, bei
Literatur Kasten
denen die DNA-Sequenz irreversibel verändert wird, kommt es beim Imprinting nur zu einer reversiblen und funkti-
a. Cattanach DM et al.: onellen Modifikation der DNA, bei der die DNA-Sequenz selbst unverändert bleibt. Bei einem maternalen Imprin-
Differential acivity of mater-
nally and paternally ting ist das mütterliche Allel inaktiv, während das väterliche Allel exprimiert wird; bei einem paternalen Imprinting
derived chromosome regi- verhält es sich genau umgekehrt. Genomisches Imprinting bedarf bestimmter epigenetischer Marker, mit deren
ons in mice. Nature. 1985; Hilfe die transkriptionale Aktivität bestimmter Genombereiche reguliert wird. Dabei ist vor allem die differenzielle
315(6019): 496-498
DNA-Methylierung (Anhängen einer CH3-Gruppe an Cytosinreste) gut untersucht [b]. Des Weiteren werden Histone
b. Bird AP: CpG-rich islands
and the function of DNA und kleine, regulatorische RNA-Moleküle als mögliche epigenetische Marker diskutiert [c].Veränderungen in den
methylation. Nature. 1986; epigenetischen Markern, also z.B. im Methylierungsmuster eines Genabschnitts, werden auch als epigenetische
321(6067): 209-213 Mutationen oder Epimutationen bezeichnet. Geprägte bzw. imprintete Gene finden sich häufig in Gen-Clustern,
c. Jaenisch R et al.: Epigen- die an sogenannten differenziert methylierten Regionen (engl.: differentially methylated regions, DMRs) erkannt
tic regulation of gene
expression: how the geno- werden können. Einige dieser DMRs werden auch als Imprintingcenter-Regionen (engl.: imprinting control region,
me integrates intrinsic and ICRs) bezeichnet. Eine Vielzahl geprägter Gene spielt eine essenzielle Rolle bei der fetalen Entwicklung. Dabei
environmental signals. wurde beobachtet, dass paternal exprimierte Allele das Wachstum erhöhen, während maternale exprimierte Allele
Nat Genet. 2003;
33 (Suppl.): 245-254 das Wachstum hemmen [d].
d. Reik W, et al.: Genomic Eine Störung im Imprinting kann unabhängig von der Ursache zu einer Reihe von Erkrankungen führen. Diese kön-
imprinting: parental influ- nen dadurch verursacht werden, dass normalerweise stumme Gene aufgrund von fehlender Prägung aktiv werden
ence on the genome. Nat und damit die Menge an Genprodukten verdoppelt wird oder dass durch Repression normalerweise aktiver Gene
Rev Genet. 2001; 2(1): 21-32
essenzielle Genprodukte fehlen. Dies kann unter anderem durch die uniparentale Disomie (UPD) geschehen. Bei
e. Kotzot D et al.: Uniparen-
tal Disomy (UPD) other than der UPD stammen beide homologen Chromosomen eines Chromosomenpaares oder einer chromosomalen Regi-
15: phenotypes and bib- on von einem einzigen Elternteil ohne Beteiligung des anderen Elternteils. Dies kann in Bereichen mit geprägten
liography updated. Am J
Genen zu einer Störung des Imprintings führen, da der biparentale Beitrag zur Genregulation fehlt. Die UPD wird
Med Genet A. 2005; 136(3):
287-305 mittlerweile mit einer Reihe von Störungen in Verbindung gebracht: Prader-Willi-Syndrom (maternal UDP15), An-
gelman-Syndrom (paternal UDP15), Beckwith-Wiedemann-Syndrom (paternal UDP11p15.5) und das Silver-Russell-
Syndrom (maternal UDP7) [e].
Bei dem Silver-Russell-Syndrom (SRS, auch RSS, rogenität der klinischen Symptome macht die Ab-
Russell-Silver-Syndrom; OMIM 180860) handelt es grenzung und genaue Diagnostik vom SRS gegen-
sich um eine seltene, klinisch heterogene Erkran- über anderen Wachstumsstörungen schwierig, auch
kung, die durch prä- und postnatale Wachstumsstö- wenn mittlerweile Leitsymptome und diagnostische
rungen, typische Gesichtsdysmorphien und laterale Kriterien für die Erkrankung postuliert wurden [3].
Wachstumsasymmetrien charakterisiert wird. Silver Erschwerend kommt hinzu, dass sich die klinische
(1953) und Russell (1954) beschrieben unabhängig Symptomatik mit zunehmendem Alter abschwächt.
voneinander die Kleinwüchsigkeit bei Neugebore- Aus diesem Grund sind Angaben zur Prävalenz vom
nen und Kleinkindern, eine typische dreieckige Ge- SRS schwierig; die Zahlen schwanken zwischen ei-
sichtsform mit einer breiten Stirn und einem spitzen nem und neun Fällen auf 100 000 [4].
Kinn, einen normalen Kopfumfang trotz Minder-
wuchs, tief angesetzte Ohren und Anomalien der
Genetik
Extremitäten, wie die Kurzfingrigkeit [1,2]. Mitt- Neben der großen klinischen Heterogenität zeigt
lerweile sind eine Reihe von weiteren Symptomen sich das SRS auch hinsichtlich der Krankheitsursa-
wie Hypogonadismus, übermäßiges Schwitzen und chen sehr heterogen. Von den mittlerweile über
Schwierigkeiten bei der Ernährung der Babys und 400 in der medizinischen Literatur beschriebenen
Kleinkinder hinzugekommen [3]. Die große Hete- Fällen sind die meisten zwar sporadischen Ur-
11SR S
[8]. Weitergehende Untersuchungen identifizier-
ten zwei separate Regionen auf Chromosom 7 mit
einer Reihe von geprägten Genen, auf die eine
mUPD Auswirkungen haben könnte: die Region
7p11.2-7p13 und die Region beginnend bei 7q31
(Abbildung 1). Beide Regionen sind homolog zu
bestimmten geprägten Chromosomenregionen
bei Mäusen, die dort alle mit Wachstumsphänoty-
pen assoziiert sind [9]. So wurden Wachstumsver-
zögerungen und Insulinresistenz bei Mäusen mit
Überexpression in demjenigen Gen beobachtet,
das dem humanen Gen GRB10 in der Chromoso-
menregion 7p11.2-7p13 homolog ist. Beim Men-
schen kodiert GRB10 (engl. growth factor recep-
tor-bound protein 10) für ein zytoplasmatisches
Adapterprotein, das mit Tyrosinkinase-Rezeptoren
interagiert, u.a. auch mit dem IGF-1-Rezeptor. Zu
diesen Beobachtungen bei Mäusen passten die
Befunde dreier SRS-Patienten mit typischem Phä-
notyp und einer Verdopplung im 7p-Segment und
damit von GRB10, womit der beobachtete Minder-
wuchs analog zu den Beobachtungen bei Mäusen
durch die Überexpression von GRB10 zu erklären
Abbildung 1: sprungs, es gibt allerdings auch einige Fälle mit
wäre. Es wurde postuliert, dass auch eine mUPD7
SRS-Kandidatengene auf familiärem Hintergrund. Dabei wurden sowohl
Chromosom 7 zu einer Überexpression von GRB10 und damit zu
X-chromosomale als auch autosomal-rezessive
MEST=mesoderm specific Minderwuchs bei den betroffenen Patienten führen
transcript und autosomal-dominante Vererbungsmuster be-
müsste. Doch trotz umfassender Screenings bei
CPA4=carboxypeptidase A4 schrieben [3]. Im Jahr 1995 wurde zudem der Fall
COPG2=coatomer protein SRS-Patienten mit mUPD7 konnten bis jetzt keine
von eineiigen Zwillingen veröffentlicht, bei dem
complex subunit gamma 2 Mutationen oder epigenetischen Aberrationen im
MESTIT=MEST intronic tran- nur ein Zwilling vom SRS betroffen war [5]. Dies
script, non protein coding
Methylierungsmuster von GRB10 gefunden wer-
führte mit den ersten Fällen von maternaler unipa-
CIT1=intronic transcript, den. Somit ist der mit einer mUPD7 assoziierte
non protein coding rentaler Disomie (mUPD) auf Chromosom 7 beim
pathogene Mechanismus, der zum SRS führt, nach
IGFBP1, 3=insulin-like SRS [6] zu der Hypothese, dass vor allem epige-
growth factor binding pro- wie vor ungeklärt und ein deutlicherer Effekt von
netische Veränderungen ursächlich für das SRS
tein 1 or 3 GRB10 auf den mUPD7-SRS-Phänotyp wird mittler-
GRB10=growth factor re- sein könnten. In den folgenden Jahren wurde eine
ceptor-bound protein 10
weile ausgeschlossen [3, 10]. Trotzdem bleibt die
Vielzahl von Kandidatengenen auf unterschiedli-
EGFR=epidermal growth Region 7p11.2-7p13 für die Wissenschaft weiter
factor receptor chen Chromosomen diskutiert, wobei die beiden
interessant, da sich dort mit IGFBP1 und IGFBP3
*geprägte Gene vielversprechendsten Kandidaten die Chromoso-
noch die Gene für zwei Bindungsproteine von IGF
Quelle: modifiziert nach [11]
men 7 und 11 sind. Genetische wie epigenetische
(engl. insulin-like growth factor) sowie mit EGFR
Veränderungen auf diesen beiden Chromosomen
das Gen für den EGF-Rezeptor (eng. epidermal
werden mittlerweile bei bis zu 70 Prozent aller SRS-
growth factor) befinden (Abbildung 1).
Patienten gefunden [7].
Ähnlich sieht es auch für die zweite Kandida-
tenregion auf dem langen Arm des Chromosoms
Häufige Beteiligung von Chromosom 7 7 aus. Vier Patienten mit verzögertem Wachstum
Routinemäßige Untersuchungen der letzten Jahre und einer segmentalen mUPD im Bereich 7q31
ergaben, dass etwa sieben bis zehn Prozent aller sind mittlerweile in der Literatur bekannt, drei da-
SRS-Patienten von einer mUPD7 betroffen sind von weisen typische SRS-Merkmale auf. Auch in
12SR S
dieser chromosomalen Region befinden sich mit ein Cluster von weiteren geprägten Genen, die eine
MEST, CPA4 und COPG2 drei geprägte Gene und wichtige Rolle beim fetalen Wachstum spielen. Die-
mit MESTIT und CIT1 zwei nicht proteinkodieren- ses Cluster enthält neben dem paternal exprimierten
Abbildung 2:
Regulation in der telome- de RNAs (Abbildung 1). Allerdings haben Scree- Gen IGF-2 noch das ebenfalls paternal exprimierte
ren Region von 11p15 bei nings auch hier noch keine pathogenen Varianten KCNQ1OT1 sowie die maternal exprimierten Gene
normalen Individuen und
ergeben, sodass ein Einfluss auf den SRS-Phäno- CDKN1C und H19. Die geprägte Region besteht
Patienten mit SRS (Silver-
Russell-Syndrom) und BWS typ ebenfalls fraglich ist [11]. aus zwei Imprintingdomänen, einer zentromeren
(Beckwith-Wiedemann-
und einer telomeren, die jeweils unter der Kontrolle
Syndrom) durch differen-
zierte Methylierung Die Rolle des Chromosoms 11 ihrer eigenen Imprintingcenter-Region (ICR) stehen
IGF-2=insulin-like growth
IGF-2 spielt beim Menschen eine kritische Rolle in (Abbildung 2). Die telomere ICR1 reguliert dabei die
factor 2
der fetalen Entwicklung und wird vor allem mono- beiden geprägten Gene IGF-2 und H19, während
ICR1, ICR2=Imprinting-
center-Region 1 oder 2 allelisch über die Promotoren P3 und P4 abhängig die Expression von KCNQ1OT1 und CDKN1C von
H19=imprinted maternally der zentromeren ICR2 reguliert wird [14].
expressed transcript (non-
von dem jeweiligen Gewebe und dem jeweiligen
protein coding) Entwicklungsstadium exprimiert [12]. Störungen in Aufgrund der wichtigen Rolle, die IGF-2 beim
CDKN1C=cyclin-dependent fetalen Wachstum spielt, wurde besonders die Re-
der IGF-2-Expression während der Fetalentwicklung
kinase inhibitor 1c
KCNQ1=potassium voltage- führen zu intrauterinen Wachstumsveränderungen. gion um den ICR1 genauer untersucht [15]. Dabei
gated channel KQT-Subfa- Das Gen für IGF-2 befindet sich beim Menschen in wurde deutlich, dass die wechselseitige Expression
mily member 1
der geprägten Genregion 11p15, für die wiederholt vom maternal exprimierten H19 und dem paternal
KCNQ1OT1=KCNQ1
overlapping transcript 1 maternale Duplikationen bei Patienten mit klinisch ty- exprimierten IGF-2 auf einer differenzierten Methy-
(non-protein coding)
pischen SRS-Merkmalen – Minderwuchs, dreieckiges lierung der ICR1 beruht. Auf den maternalen Chro-
CTCF :CCCTC-binding
factor Gesicht, normaler Kopfumfang – beschrieben wurden mosomen ist die ICR1 nicht methyliert und wird so
Quelle: modifiziert nach [3] [7,13]. Neben IGF-2 befindet sich im Bereich 11p15 von CTCF, einem Zink-Finger-Protein, gebunden.
13SR S
7. Eggermann T et al.: Is maternal Durch diese Verbindung der ICR1 mit CTCF bildet Gene, also durch eine IGF-2-Überexpression, zu
duplication of 11p15 associated
with Silver-Russell syndrome? J sich eine Art Isolator, der die Interaktion der dem erklären ist. Dagegen werden beim SRS mit dem
Med Genet. 2006; 42(5): e26
H19 nachfolgenden Enhancer mit dem Promotor klinischen Bild des Minderwuchses vermehrt mater-
8. Kotzot D et al.: Maternal unipa-
rental disomy 7: review and further des IGF-2-Gens unterbindet (Abbildung 2a) [16, nale und wachstumshemmende Gene exprimiert
delineation of the phenotype. Eur
J Ped. 2000; 159(4): 247-256 17]. Im Gegensatz dazu ist die ICR1 des paternalen beziehungsweise wachstumsfördernde Gene wie
9. Miyoshi N et al.: Identification Chromsomens methyliert, was eine Bindung von IGF-2 unterdrückt. Durch den Vergleich der beiden
of the mMeg1/Grb10 imprinted
gene on mouse proximal chro- CTCF an die Imprintingcenter-Region verhindert. Syndrome wird deutlich, dass epigenetische Verän-
mosome 11, a candidate for the
Silver-Russell syndrome gene. Somit kann der H19 nachgeschaltete Enhancer mit derungen in der gleichen Domäne eines Chromo-
Proc Natl Acad Sci USA 1998;
95(3): 1102-1107
dem Promotor des IGF-2-Gens interagieren und für soms ein gegensätzliches klinisches Bild hervorru-
10. Riegel M et al.: No evidence dessen Transkription sorgen. Bei Untersuchungen fen können. Während für das BWS allerdings auch
of submicroscopic deletion or
segmental uniparental disomy einer Patientengruppe von neun typischen SRS- die zentromere Domäne eine Rolle spielt, sind für
within the candidate regions das SRS für diese Domäne bis jetzt noch keine Epi-
Patienten mit einer biparentalen Vererbung in dem
7p11.2-p13 and 7q31-qter in a
series of non-uniparental disomy 11p15-Bereich wurde bei fünf Patienten ein partiel- mutationen von Relevanz nachgewiesen worden.
Silver-Russell syndrome cases.
Clin Genet. 2003; 64(3): 252-254 ler Verlust der Methylierung (LOM, loss of methyla-
11. Eggermann T: Epigenetic tion) in der ICR1 beobachtet [15]. Resultat dieser Ausblick
regulation of growth: lessons
from the Silver-Russell syndrome. Hypomethylierung ist eine Bindung von CTCF an
Endocr Dev. 2009; 14: 10-19 Da SRS nicht nur aufgrund von Veränderungen
die ICR1 und eine Abschirmung und damit Unter-
12. Wu HK et al.: Promoter-de- in den Chromosomen 7 und 11 auftritt, sondern
pendent tissue-specific expres- drückung der Transkription des IGF-2-Gens auch
sive nature of imprinting gene, auch bei veränderten Chromosomen 15 und 17
insulin-like growth factor II, in auf dem paternalen Chromatiden. Somit wird H19
human tissues. Biochem Biophys
beschrieben wurde, wird es bis zur Aufklärung der
biallelisch exprimiert, während eine IGF-2-Expres-
Res. 1997; 233(1): 221-226 molekularen Mechanismen des SRS noch einiger
13. Fisher AM et al.: Duplications sion nicht mehr stattfindet (Abbildung 2b). Weitere
of chromosome 11p15 of maternal Zeit bedürfen. Trotz der großen genetischen Hete-
origin result in a phenotype that Untersuchungen bestätigten diese Beobachtungen
rogenität fällt jedoch auf, dass bei den Chromso-
includes growth retardation. Hum
auch in anderen Patientengruppen [18].
Genet. 2002; 111(3): 290-296 men 7 und 11 (und auch bei Patienten mit Anoma-
14. Weksberg R et al.: Beckwith-
lien in Chromosomen 15) in irgendeiner Weise das
Wiedemann syndrome demonst-
rates a role for epigenetic control
Hypomethylierung und Schweregrad IGF-System betroffen zu sein scheint. In einer ak-
of normal development. Hum
Mol Genet. 2003; 12(1): R61-R68
der Erkrankung tuellen Veröffentlichung postuliert Eggermann da-
15. Gicquel C et al.: Epimutation
Neueste Forschungsergebnisse beschreiben dar- her, dass das SRS durch Veränderungen in einem
of the telomeric imprinting center
region on chromosome 11p15 über hinaus einen Zusammenhang zwischen dem oder mehreren Faktoren entlang der IGF-Achse in
in Silver-Russell syndrome. Nat
Genet. 2005; 37(9): 1003-1007 Grad der Hypomethylierung an der ICR1 und der unserem Körper verursacht wird [11]. Ein genaue-
16. Bell AC, Felsenfeld G: Me- Schwere des SRS-Phänotyps [19]. Dabei scheinen res Verständnis dieser Störungen im IGF-Netzwerk
thylation of a CTCF-dependent
boundary controls imprinted besonders orthopädische Anomalien der Hände, könnte in der Zukunft eine zielgerichtetere Thera-
expression of the Igf2 gene. Na-
ture. 2000; 405(6785): 482-485 Füße und der Wirbelsäule bei SRS-Patienten mit ex- pie mit Wachstumshormonen ermöglichen.
17. Hark AT et al.: CTCF tremer Hypomethylierung in der ICR1 aufzutreten,
mediates methylation-sensitive Dr. Eva A. Schulte
enhancer-blocking activity at the die bei Patienten mit gering hypomethylierter ICR1
H19/Igf2 locus. Nature. 2000; Literatur
405(6785): 486-489
deutlicher weniger auffällig sind. Aufschlussreich
1. Silver HK et al.: Syndrome of congential hemihyperthrophy,
18. Eggermann T et al.: Epigenetic ist auch ein Vergleich des SRS mit dem Beckwith-
shortness of stature, and elevated urinary gonadotropins. Pe-
mutations in 11p15 in Silver-Russell
syndrome are restricted to the telo-
Wiedemann-Syndrom (BWS), das unter anderem diatrics. 1953; 12(4): 368-376
2. Russell A: A syndrome of intra-uterine dwarfism recognizable
meric imprinting domain. J Med durch Großwuchs charakterisiert ist [20]. Eine Reihe
Genet. 2006; 43(7): 615-616 at birth with cranio-facial dysostosis, disproportionately short
19. Bruce S et al.: Clinically von chromosomalen Anomalien wurde für das BWS arms, and other anomalies (5 examples). Proc R Soc Med.
distinct epigenetic subgroups
beschrieben, unter anderem auch für die zentrome- 1954; 47(12): 1040-1044
in Silver-Russell syndrome: the 3. Rossignol S et al.: Epigenetics in Silver-Russell syndrome. Best
degree of H19 hypomethylati- ren und telomeren Imprintingdomänen der 11p15- Pract Res Clin Endocrinol Metab. 2008; 22(3): 403-414
on associates with phenotype
severity and genital and skeletal Region. Beim BWS wurde im Gegensatz zum SRS 4. www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=DE&Expert
anomalies. J Clin Endocrinol =813
Metab. 2009; 94(2): 579-587 allerdings eine Hypermethylierung der ICR1 beob-
5. Bailey W et al.: Monozygotic twins discordant for Russell-Sil-
20. Eggermann T et al.: Growth achtet, sodass auch bei dem maternalen Chroma- ver syndrome. Am J Med Genet. 1995; 58(2): 101-105
retardation versus overgrowth: 6. Kotzot D et al.: Uniparental Disomy 7 in Silver-Russell syndro-
Silver-Russell syndrome is tiden IGF-2 exprimiert wurde (Abbildung 2c). Dies
me and primodial growth retardation. Hum Mol Genet. 1995;
genetically opposite to Beckwith-
Wiedemann syndrome. Trends
passt zu dem klinischen Bild des Großwuchses, das 4(4): 583-587
Genet. 2008; 24(4): 195-204 durch paternal exprimierte, wachstumsfördernde Fortsetzung Literatur siehe linke Spalte
14Sie können auch lesen

















































